
AutoDock4 and AutoDock Vina are the most commonly used open-source software for protein-ligand docking. However, they both rely on a derivative of the “PDB” (Protein Data Base) file format: the “PDBQT” file (Protein Data Bank, Partial Charge (Q), & Atom Type (T)). In addition to the information contained in normal PDB files, PDBQT files have an additional column that lists the partial charge (Q) and the assigned AutoDock atom type (T) for each atom in the molecule. AutoDock atom types offer a more granular differentiation between atoms such as listing aliphatic carbons and aromatic carbons as separate AutoDock atom types.
The biggest drawback about the PDBQT format is that it does not encode for the bond order in molecules explicitly. Instead, the bond order is inferred based on the atom type, distance and angle to nearby atoms in the molecule. For normal sp3 carbons and molecules with mostly single bonds this system works fine, however, for more complex structures containing for example aromatic rings, conjugated systems and hypervalent atoms such as sulphur, the bond order is often not displayed correctly. This leads to issues downstream in the screening pipeline when molecules suddenly change their bond order or have to be discarded after docking because of impossible bond orders.
The solution to this problem is included in RDKit: The AssignBondOrdersFromTemplate function. All you need to do is load the original molecule used for docking as a template molecule and the docked pose PDBQT file into RDKIT as a PDB, without the bond order information. Then assign the original bond order from your template molecule. The following code snippet covers the necessary functions and should help you build a more accurate and reproducible protein-ligand docking pipeline:
#import RDKit AllChem from rdkit import Chem from rdkit.Chem import AllChem #load original molecule from smiles SMILES_STRING = "CCCCCCCCC" #the smiles string of your ligand template = Chem.MolFromSmiles(SMILES_STRING) #load the docked pose as a PDB file loc_of_docked_pose = "docked_pose_mol.pdb" #file location of the docked pose converted to PDB file docked_pose = AllChem.MolFromPDBFile(loc_of_docked_pose) #Assign the bond order to force correct valence newMol = AllChem.AssignBondOrdersFromTemplate(template, docked_pose) #Add Hydrogens if desired. "addCoords = True" makes sure the hydrogens are added in 3D. This does not take pH/pKa into account. newMol_H = Chem.AddHs(newMol, addCoords=True) #save your new correct molecule as a sdf file that encodes for bond orders correctly output_loc = "docked_pose_assigned_bond_order.sdf" #output file name Chem.MolToMolFile(newMol_H, output_loc)

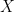 into two sets – a training set
into two sets – a training set 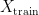 and a test set
and a test set 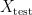 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 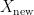 which it will encounter in the future. Right?
which it will encounter in the future. Right?

