
We have reached the era of design, not just ‘hunting’. Particularly exciting to me is the de novo design of proteins, which have a wide and ever increasing range of applications from therapeutics to consumer products, biomanufacturing to biomaterials. Protein design has been a) enabled by decades of research that contributed to our understanding of protein sequence, structure & function and b) accelerated by computational advances – capturing the information we have learned from proteins and representing it for computers and machine learning algorithms.
In this blog post, I will discuss three key methodological considerations for computational protein design:
- Sequence- vs structure-based design
- ML- vs physics-based design
- Target-agnostic vs target-aware design

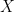 into two sets – a training set
into two sets – a training set 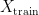 and a test set
and a test set 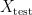 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 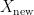 which it will encounter in the future. Right?
which it will encounter in the future. Right?