
Recently, I have been interested in adding a confidence metric to the predictions made by a machine learning model I have been working on. In this blog post, I will outline a few strategies I have been exploring to do this. Powerful deep learning models like AlphaFold are great, not only for the predictions they make, but they also generate confidence measures to give the user a sense of how much to trust the prediction.
Continue readingTag Archives: machine learning
Estimating the Generalisability of Machine Learning Models in Drug Discovery
Machine learning (ML) has significantly advanced key computational tasks in drug discovery, including virtual screening, binding affinity prediction, protein-ligand structure prediction (co-folding), and docking. However, the extent to which these models generalise beyond their training data is often overestimated due to shortcomings in benchmarking datasets. Existing benchmarks frequently fail to account for similarities between the training and test sets, leading to inflated performance estimates. This issue is particularly pronounced in tasks where models tend to memorise training examples rather than learning generalisable biophysical principles. The figure below demonstrates two examples of model performance decreasing with increased dissimilarity between training and test data, for co-folding (left) and binding affinity prediction (right).

A tougher molecular data split – spectral split
Scaffold splits have been widely used in molecular machine learning which involves identifying chemical scaffolds in the data set and ensuring scaffolds present in the train and test sets do not overlap. However, two very similar molecules can have differing scaffolds. In an example provided by Pat Walters in his article on splitting chemical data last month, he provides an example where two molecules just differ by a single atom and thus have a very high Tanimoto similarity score of 0.66. However, they have different scaffolds (figure below).

In this case, if one of the molecules were in the train set and the other in the test set, predicting the test molecule would be quite trivial as there is data leakage. Therefore, we need a better splitting method such that there is minimal overlap between the train and test set. In this blogpost, I will be discussing spectral split, a splitting method introduced by our fellow OPIG member, Klarner et. al (2023).
Spectral split
Spectral split or clustering is based on the spectral graph partitioning algorithm. The basic idea of spectral clustering is as follows: The dataset is projected on a R^n matrix. An affinity matrix using a kernel that could be domain-specific is defined. Following that, the graph Laplacian is computed from the affinity matrix, followed by its eigendecomposition. Then, k eigenvectors corresponding to the k lowest/highest eigenvalues are selected. Finally, the clusters are formed using k-means.
In the context of molecular data splitting, one could use the Tanimoto similarity metric to construct a similarity matrix between all the molecules in the dataset. Then, a spectral clustering method could be used to partition the similarity matrix such that the similarity within the cluster is maximized whereas the similarity between the clusters is minimized. Spectral split showed the least overlap between train (blue) and test (red) set molecules compared to scaffold splits (figure from Klarner at. al. (2024) below)

In addition to spectral splits, one could attempt other tougher splits one could attempt such as UMAP splits suggested by Guo et. al. (2024). For a detailed comparison between UMAP splits and other commonly used splits please refer to Pat Walters’ article on splitting chemical data.
Visualising and validating differences between machine learning models on small benchmark datasets
Introduction
An epidemic is sweeping through cheminformatics (and machine learning) research: ugly results tables. These tables are typically bloated with metrics (such as regression and classification metrics next to each other), vastly differing tasks, erratic bold text, and many models. As a consequence, results become difficult to analyse and interpret. Additionally, it is rare to see convincing evidence, such as statistical tests, for whether one model is ‘better’ than another (something Pat Walters has previously discussed). Tables are a practical way to present results and are appropriate in many cases; however, this practicality should not come at the cost of clarity.
The terror of ugly tables extends to benchmark leaderboards, such as Therapeutic Data Commons (TDC). These leaderboard tables do not show:
- whether differences in metrics between methods are statistically significant,
- whether methods use ensembles or single models,
- whether methods use classical (such as Morgan fingerprints) or learned (such as Graph Neural Networks) representations,
- whether methods are pre-trained or not,
- whether pre-trained models are supervised, self-supervised, or both,
- the data and tasks that pre-trained models are pre-trained on.
This lack of context makes meaningful comparisons between approaches challenging, obscuring whether performance discrepancies are due to variance, ensembling, overfitting, exposure to more data, or novelties in model architecture and molecular featurisation. Confirming the statistical significance of performance differences (under consistent experimental conditions!) is crucial in constructing a more lucid picture of machine learning in drug discovery. Using figures to share results in a clear, non-tabular format would also help.
Statistical validation is particularly relevant in domains with small datasets, such as drug discovery, as the small number of test samples leads to high variance in performance between different splits. Recent work by Ash et al. (2024) sought to alleviate the lack of statistical validation in cheminformatics by sharing a helpful set of guidelines for researchers. Here, we explore implementing some of the methods they suggest (plus some others) in Python.
Continue readingCream, Compression, and Complexity: Notes from a Coffee-Induced Rabbit Hole
I have recently stumbled upon this paper which, quite unexpectedly, sent me down a rabbit hole reading about compression, generalisation, algorithmic information theory and looking at gifs of milk mixing with coffee on the internet. Here are some half-processed takeaways from this weird journey.
Complexity of a cup of Coffee
First, check out this cool video.
Maybe we should train on our test set?
One of the fundamental (pitfalls) of machine learning is to ensure that you don’t train on your test set, but what if I told you that you could?

Conference summary: Generative AI in Life Science
This year I attended the second edition of Generative AI in Life Science (GenLife – https://genlife.dk/) and it was an enriching experience that I thoroughly enjoyed. Held in Copenhagen, the event brought together researchers from different areas of AI applied to the life sciences and provided a fantastic platform for networking, learning and sharing ideas. The programme included a mix of long and short talks from experts in the field, but also had a significant presence of emerging PIs, making the conference a perfect place to discover emerging groups in the field. Here I have collected some highlights of the talks I have enjoyed the most at the conference.
Continue readingUsing JAX and Haiku to build a Graph Neural Network
JAX
Last year, I had an opportunity to delve into the world of JAX whilst working at InstaDeep. My first blopig post seems like an ideal time to share some of that knowledge. JAX is an experimental Python library created by Google’s DeepMind for applying accelerated differentiation. JAX can be used to differentiate functions written in NumPy or native Python, just-in-time compile and execute functions on GPUs and TPUs with XLA, and mini-batch repetitious functions with vectorization. Collectively, these qualities place JAX as an ideal candidate for accelerated deep learning research [1].
JAX is inspired by the NumPy API, making usage very familiar for any Python user who has already worked with NumPy [2]. However, unlike NumPy, JAX arrays are immutable; once they are assigned in memory they cannot be changed. As such, JAX includes specific syntax for index manipulation. In the code below, we create a JAX array and change the 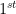
Tracking the change in ML performance for popular small molecule benchmarks
The power of machine learning (ML) techniques has captivated the field of small molecule drug discovery. Increasingly, researchers and organisations have employed ML to create more accurate algorithms to improve the efficiency of the discovery process.
To be published, methods have to prove they have improved upon others. Often, methods are tested against the same benchmarks within a field, allowing us to track progress over time. To explore the rate of improvement, I curated the performance on three popular benchmarks. The first benchmark is CASF 2016, used to test the accuracy of methods that predict the binding affinity of experimental determined protein-ligand complexes. Accuracy was measured using the Pearson’s R value between predicted and experimental affinity values.
Continue readingOptimising for PR AUC vs ROC AUC – an intuitive understanding
When training a machine learning (ML) model, our main aim is usually to get the ‘best’ model out the other end in an unbiased manner. Of course, there are other considerations such as quick training and inference, but mostly we want to be good at predicting the right answer.
A number of factors will affect the quality of our final model, including the chosen architecture, optimiser, and – importantly – the metric we are optimising for. So, how should we pick this metric?
Continue reading