
This time round, one older paper, one recent paper. The older one talks about estimating how many H3s can there be in a human body based on sequencing of two individuals (they cap it at 9 million — not that much!). The more recent one is an attempt to define what makes a good antibody in terms of its developability properties (a battery of biophys assays on 150 therapeutic antibodies- amazing dataset to work with).
High resolution description of antibody heavy chain repertoires in (two) humans (Koralov lab at NYU). Here. Two individuals were sequenced and their VDJ frequencies measured. It is widely believed that the VDJ recombination events are largely independent and random. Here however they demonstrate some biases/interplay between the D and J regions. Since H3 falls on the VDJ junction, it might suggest that it affects the total choice of H3. Another quite important point is that they compared the productive vs nonproductive sequences (out of frame or with stop codons). If there were significant differences between the VDJ frequencies of productive vs nonproductive sequences, it would suggest selection at the VDJ frequency stage. However they do not see any significant differences here suggesting that VDJ combinations have little bearing on this initial selection step. Finally, they estimate the number of H3 in repertoire. The technique is interesting — they sample 1000 H3s from their set and see how many unique sequences it contributes. Each next sample contributes less and less unique sequences which leads to a log-decay curve. By doing so they get a rough estimate of when there will be no more new sequences added and hence an estimate of diversity (think why do this rather than counting the number of uniques!). They allow themselves to extrapolate this estimate to the whole organism by multiplying their blood sample by the total human body volume — they motivate this extrapolation by the fact that there were precious little overlaps between the two human subjects.
Biophysical landscape of clinical stage antibodies [here]. Paper from Adimab. Designing an antibody which binding its target is only a first step on the way to bring the drug on the market. The molecule needs to fulfill a variety of characteristics such as colloidal stability (does not aggregate or ‘clump up’), does not instantly clear from the organism (which is usually down to off target binding), is stable and can be expressed in reasonable quantities. In an effort to delineate what makes a good antibody, the authors take inspiration from earlier work on small molecules, namely the Lipinski Rules of Five. This set of rules describes what makes a ‘good’ small molecule drug, which was assessed by looking at ~2000 therapeutic drugs. The rules came down to certain numbers of hydrogen bond donors, acceptors, molecular weight & lipophilicity. Therefore, Jain et al would like a similar methodology, but for antibodies: give me an antibody and using methodology/rules we define, we will tell you either to carry on with development or maybe not. Since antibodies are far more complex and the data on therapeutic abs orders of magnitude smaller (around 50 therapeutic abs to date) Jain et al, had to devise a more nuanced approach than simply counting hb donors/acceptors mass etc. The underlying ‘good’ molecule data though is similar: they picked therapeutic antibodies and those in late clinical testing stages (2,3). This resulted in ~150 antibodies. So as to devise the benchmark ‘rules/methodology’, they went for a battery of assays to serve as a benchmark — if your ab raises too many red flags according to these assays, it’s not great (what constitutes a red flag to be defined). These assays were supposed to not be obscure and relatively easy to use as the point was that an arbitrary antibody can be relatively easy checked against them. The assays are a range of expression, cross reactivity, self reactivity, thermal stability etc. To define red flags, they run their therapeutic/clinical antibodies through the tests. To their surprise quite a lot of these molecules turn out to have quite ‘undesirable characteristics’. Following the Lipinski Rules, they define a red flag as being in the bottom 10th percentile of the assay values as evaluated on the therapeutic abs. They show that the antibodies which are approved or in more advanced clinical trials stages have less red flags. Therefore, the take-home messages from this paper: very nice dataset for any computational work, raising red flags does not disqualify you from being a therapeutic.






 or equivalently with their dispersion
or equivalently with their dispersion  .
. , where
, where 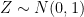 is an error term,
is an error term, 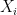 its the value recorded by instrument
its the value recorded by instrument 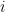 and where
and where 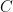 is the fixed true quantity of interest the instrument is trying to measure. But, what if
is the fixed true quantity of interest the instrument is trying to measure. But, what if  . Therefore the instruments would record quantities of the form
. Therefore the instruments would record quantities of the form 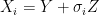 .
.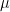 , the expected state of the phenomenon of interest is not a big challenge. Assume that there are
, the expected state of the phenomenon of interest is not a big challenge. Assume that there are  values observed from realisations of the variables
values observed from realisations of the variables 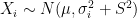 , which came from
, which came from 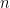 different instruments. Here
different instruments. Here 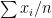 is still a good estimation of
is still a good estimation of 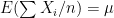 . Now, a more challenging problem is to infer what is the underlying variability of the phenomenon of interest
. Now, a more challenging problem is to infer what is the underlying variability of the phenomenon of interest  . Under our previous setup, the problem is reduced to estimating
. Under our previous setup, the problem is reduced to estimating  as we are assuming
as we are assuming 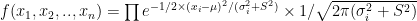 ,
,![\sum [(X_i- \mu)^2 - (\sigma_i^2 + S^2)] /(\sigma_i^2 + S^2)^2 = 0](https://s0.wp.com/latex.php?latex=%5Csum+%5B%28X_i-+%5Cmu%29%5E2+-+%28%5Csigma_i%5E2+%2B+S%5E2%29%5D+%2F%28%5Csigma_i%5E2+%2B+S%5E2%29%5E2+%3D+0&bg=ffffff&fg=000&s=0&c=20201002) .
.![E[\sum (X_i-\sum X_i/n)^2] = (1-1/n) \sum \sigma_i^2 + (n-1) S^2](https://s0.wp.com/latex.php?latex=E%5B%5Csum+%28X_i-%5Csum+X_i%2Fn%29%5E2%5D+%3D+%281-1%2Fn%29+%5Csum+%5Csigma_i%5E2+%2B+%28n-1%29+S%5E2&bg=ffffff&fg=000&s=0&c=20201002)
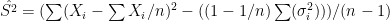 .
. and where the variance of the phenomenon of interest
and where the variance of the phenomenon of interest  . In the first scenario
. In the first scenario ![[10,1500^2]](https://s0.wp.com/latex.php?latex=%5B10%2C1500%5E2%5D&bg=ffffff&fg=000&s=0&c=20201002) , in the second from
, in the second from ![[10,1500^2 \times 3]](https://s0.wp.com/latex.php?latex=%5B10%2C1500%5E2+%5Ctimes+3%5D&bg=ffffff&fg=000&s=0&c=20201002) and in the third from
and in the third from ![[10,1500^2 \times 5]](https://s0.wp.com/latex.php?latex=%5B10%2C1500%5E2+%5Ctimes+5%5D&bg=ffffff&fg=000&s=0&c=20201002) . In each scenario the value of
. In each scenario the value of 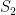 is estimated 1000 times taking each time another 200 realisations of
is estimated 1000 times taking each time another 200 realisations of 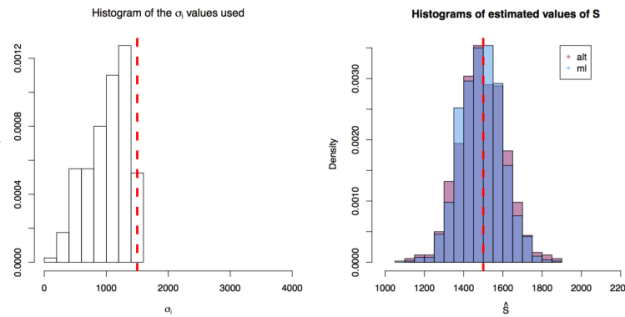 First simulation scenario where
First simulation scenario where  are shown by the blue (maximum likelihood) and red (alternative) histograms.
are shown by the blue (maximum likelihood) and red (alternative) histograms.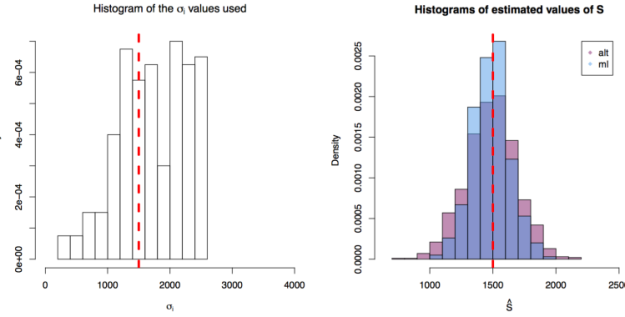 First simulation scenario where
First simulation scenario where  First simulation scenario where
First simulation scenario where 
