
Many of us in the group use python as our primary programming language. It is in my opinion an awesome language for lots of reasons. However what happens when you write an application and want to share it with the world? Simply distributing the source code requires a great deal of configuration by the end user. I’m sure you’ve all been there, you have version 1.5.1 they use version 1.6.3. However to download and install this breaks every other bit of code you are using. Creating virtual environments can help towards this, but then do you really want to go towards all the hassle of this for every application you want to use? In the end I have given up trying on a number of projects, which is a fate you would never want for your own code!
From my point of view there are three ways of counteracting this issue.
- Make limited use of libraries and imports
- Have incredibly clear instructions on how to set up the virtual env
- Freeze your code!
The first solution is sometimes just not possible or desirable. For example if you want to use a web framework or connect to third party database engines. The second could be massively time consuming and it is virtually impossible to cover all bases. For example, RDKit, my favourite cheminformatics package, has a lengthy install process with platform specific quirks and many of its own dependencies.
In my project I opted for solution number three. I use PyInstaller however there are many others available (cx_freeze, py2app, py2exe). I used PyInstaller because my application uses the Django project and they offer extra Django support. Also PyInstaller is cross-platform, allowing me (in theory) to package applications for Windows, Mac and Linux using the same protocol.
Here I will briefly outline how to set freeze your code using PyInstaller. This application validates a smiles string and shows you the RDKit canonical form of the smiles string.
This is the structure of the code:
src/
main.py
module/
__init__.py
functions.py
build/
dist/
main.py is:
import sys from module.functions import my_fun if len(sys.argv) > 1: smiles = sys.argv[1] print my_fun(smiles) else: print "No smiles string requested for validation"
functions.py is:
from rdkit import Chem
def my_fun(smiles):
mol = Chem.MolFromSmiles(smiles)
if mol is None:
return "Invalid smiles"
else:
return "Valid smiles IN: " + smiles + " OUT: " + Chem.MolToSmiles(mol,isomericSmiles=True)
- Download and install PyInstaller
- Type the following (assuming main.py is your python script)
pyinstaller src\main.py --name frozen.exe --onefile
-
This will produce a the following directory structure:
src/
main.py
module/
functions.py
build/
frozen/
dist/
frozen.exe
frozen.spec
frozen.spec is a file containing the options for building the application:
a = Analysis(['src\\main.py'],
pathex=['P:\\PATH\\TO\\HEAD'],
hiddenimports=[],
hookspath=None,
runtime_hooks=None)
pyz = PYZ(a.pure)
exe = EXE(pyz,
a.scripts,
a.binaries,
a.zipfiles,
a.datas,
name='frozen.exe',
debug=False,
strip=None,
upx=True,
console=True )
“build” contains files used in the building of the executable
“dist” contains the executable that you can distribute freely around. Because I used the “–onefile” option above it creates one single .exe file. This makes the file very easy to ship – HOWEVER for large programmes this isn’t totally ideal. All the dependencies are compressed into the .exe and uncompressed into a temporary folder at runtime. If there are lots of files, this process can be VERY slow.
So now we can run the program:
dist/frozen.exe c1ccccc1
Running dist/frozen.exe returns the error: ImportError: numpy.core.multiarray failed to import
This is because the RDKit uses this module and it is not packaged up in the frozen code. The easiest way to resolve this is to include this import in main.py:
from rdkit import Chem import numpy import sys from module.functions import my_fun if len(sys.argv) > 1: smiles = sys.argv[1] print my_fun(smiles) else: print "No smiles string requested for validation"
And there you have it. “frozen.exe” can be passed around to anyone using windows (in this case) and will work on their box.
Obviously this is a very simple application. However I have used this to package Django applications, using Tornado web servers and with multiple complex dependencies to produce native windows desktop applications. It works! Any questions, post below!










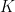 communities and that each node
communities and that each node 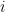 is associated with a vector of community memberships
is associated with a vector of community memberships 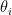 . To generate a network, the model considers each pair of nodes. For each pair
. To generate a network, the model considers each pair of nodes. For each pair 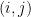 , it chooses a community indicator
, it chooses a community indicator 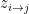 from the
from the 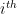 node’s community memberships
node’s community memberships 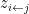 from
from  . A connection is then drawn between the nodes with probability
. A connection is then drawn between the nodes with probability 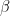 if the indicators point to the same community or with a smaller probability
if the indicators point to the same community or with a smaller probability  if they do not.
if they do not.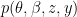 where
where 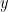 is the observed data. To estimate the posterior
is the observed data. To estimate the posterior 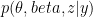 , the method uses a stochastic variational inference algorithm. This enables posterior estimation using only a sample of all-possible node pairs at each step of the variational inference, making the method applicable to very large graphs (e.g analyzing a large citation network of physics papers shown in the figure below identifies important papers impacting several sub-disciplines).
, the method uses a stochastic variational inference algorithm. This enables posterior estimation using only a sample of all-possible node pairs at each step of the variational inference, making the method applicable to very large graphs (e.g analyzing a large citation network of physics papers shown in the figure below identifies important papers impacting several sub-disciplines).


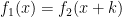 where k is the shift between the two distributions, thus if k=0 then the two populations are actually the same one. This test is based in the rank of the observations of the two samples, which means that it won’t take into account how big the differences between the values of the two samples are, e.g. if performing a WMW test comparing S1=(1,2) and S2=(100,300) it wouldn’t differ of comparing S1=(1,2) and S2=(4,5). Therefore when having a small sample size this is a great loss of information.
where k is the shift between the two distributions, thus if k=0 then the two populations are actually the same one. This test is based in the rank of the observations of the two samples, which means that it won’t take into account how big the differences between the values of the two samples are, e.g. if performing a WMW test comparing S1=(1,2) and S2=(100,300) it wouldn’t differ of comparing S1=(1,2) and S2=(4,5). Therefore when having a small sample size this is a great loss of information.