
Molecular dynamics (MD) simulations are a good way to explore the dynamical behaviour of a protein you might be interested in. One common problem is that they often have a relatively steep learning curve when using most MD engines.
What if you just want to run a simple, one-off simulation with no fancy enhanced sampling methods? OpenMM Setup is a useful tool for exactly this. It is built on the open-source OpenMM engine and provides an easy to install (via conda) GUI that can have you running a simulation in less than 5 minutes. Of course, running a simulation requires careful setting of parameters and being familiar with best practices and while this is beyond the scope of this post, there are many guides out there that can easily be found. Now on to the good stuff: using OpenMM Setup!
When you first run OpenMM Setup, you’ll be greeted by a browser window asking you to choose a structure to use. This can be a crystal structure or a model. Remember, sometimes these will have problems that need fixing like missing density or charged, non-physiological termini that would lead to artefacts, so visual inspection of the input is key! You can then choose the force field and water model you want to use, and tell OpenMM to do some cleaning up of the structure. Here I am running the simulation on hen egg-white lysozyme:



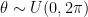 . Extending this idea to 3D rotations, we could sample each of the three Euler angles from the same uniform distribution
. Extending this idea to 3D rotations, we could sample each of the three Euler angles from the same uniform distribution 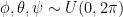 . This, however, gives more probability density to transformations which are clustered towards the poles:
. This, however, gives more probability density to transformations which are clustered towards the poles:
