Detecting intermolecular interactions is often one of the first steps when assessing the binding mode of a ligand. This usually involves the human researcher opening up a molecular viewer and checking the orientations of the ligand and protein functional groups, sometimes aided by the viewer’s own interaction detecting functionality. For looking at single digit numbers of structures, this approach works fairly well, especially as more experienced researchers can spot cases where the automated interaction detection has failed. When analysing tens or hundreds of binding sites, however, an automated way of detecting and recording interaction information for downstream processing is needed. When I had to do this recently, I used an open-source Python module called ODDT (Open Drug Discovery Toolkit, its full documentation can be found here).
My use case was fairly standard: starting with a list of holo protein structures as pdb files and their corresponding ligands in .sdf format, I wanted to detect any hydrogen bonds between a ligand and its native protein crystal structure. Specifically, I needed the number and name of the the interacting residue, its chain ID, and the name of the protein atom involved in the interaction. A general example on how to do this can be found in the ODDT documentation. Below, I show how I have used the code on PDB structure 1a9u.
Continue reading →



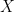 into two sets – a training set
into two sets – a training set 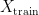 and a test set
and a test set 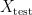 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 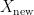 which it will encounter in the future. Right?
which it will encounter in the future. Right?

.jpg){kind=link}