
If you are as happy for the big snake as I am, you have probably wondered how you can create a Conda package with your amazing code. Fear not, in the following text you will learn how to make others go;
conda install -c coolperson amazingcodeRoughly, the only thing needed to create a Conda package, is a ‘meta.yaml’ file specific for your code. This file contains all the metadata needed to create your package and is highly customizable. While this means the meta.yaml can be written to allow your Conda package to work on any operating system and with any dependencies (doesn’t have to be python) it can be annoying to write from scratch (here is a guide for manually writing this file). Since we just want to create a simple Conda package, we will in this guide avoid fiddling around with the meta.yaml file and instead create the file based on a PyPI package. This will also give you a nice template, if you later need to adapt your meta.yaml file.
Note: Conda packages can also be made from GitHub repositories, which is likely favorable in most cases, but it also requires some manual work on the meta.yaml.


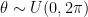 . Extending this idea to 3D rotations, we could sample each of the three Euler angles from the same uniform distribution
. Extending this idea to 3D rotations, we could sample each of the three Euler angles from the same uniform distribution 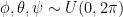 . This, however, gives more probability density to transformations which are clustered towards the poles:
. This, however, gives more probability density to transformations which are clustered towards the poles: