
Conventional wisdom dictates that compiled languages are slow to develop, can be slow to compile, but are fast to run. Interpreted languages are easy to use and do not require compilation but have sluggish performance. Like most people in scientific computing, the first two languages I learned were C++ and Python; I use Python every day but when, if ever, would I use C++?
Continue readingCategory Archives: Code
Quick Python tricks
It’s always fun when you stumble across something in your programming toolkit that you had never noticed. Here are three things I’ve recently enjoyed learning.
- Ternary syntax
a = int(raw_input()) is_even = True if a % 0 == 0 else False
- Enumerate
I’ve been looping over the length of my list, all these years, like a chump. It turns out you can do this:
for index, item in enumerate(some_list):
# now the index of each item is available as well as the item
# Don't do do this
for index in range(len(some_list)):
item = some_list[index]
- for… else
Every so often, you really need to know that a for loop has run to completion. That’s what for…else is for!
for item in iterable:
if item % 0 == 0:
first_even_number = item
else:
raise ValueError('No even numbers')
Property based testing in Python with Hypothesis : how to break your own code before someone else does
Traceback (most recent call last):
ZeroDivisionError: integer division or modulo by 0We’ve all been there. You’ve written your code, tested it out on some toy data and then when you make the move to the real data, there was something you didn’t expect.
Maybe some samples have been truncated to zero. Maybe the input arrays are the wrong shape. Suddenly your code comes crashing down around you, and you’re left thinking: well how could I have known that was going to happen? I can’t test everything
Continue readingSome useful tools
For my blog post this week, I thought I would share, as the title suggests, a small collection of tools and packages that I found to make my work a bit easier over the last few months (mainly python based). I might add to this list as I find new tools that I think deserve a shout-out.
Biopandas
Reading in .pdb files for processing and writing your own parser (while being a good exercise to familiarize yourself with the format) is a pain and clutters your code with boilerplate.
Luckily for us, Sebastian Raschka has written a neat package called biopandas [1] which enables quick I/O of .pdb files via the pandas DataFrame class.
Continue readingMaking the most of your CPUs when using python
Over the last decade, single-threaded CPU performance has begun to plateau, whilst the number of logical cores has been increasing exponentially.
Like it or loathe it, for the last few years, python has featured as one of the top ten most popular languages [tiobe / PYPL]. That being said however, python has an issue which makes life harder for the user wanting to take advantage of this parallelism windfall. That issue is called the GIL (Global Interpreter Lock). The GIL can be thought of as the conch shell from Lord of the Flies. You have to hold the conch (GIL) for your thread to be computed. With only one conch, no matter how beautifully written and multithreaded your code, there will still only be one thread will be executed at any point in time.
Automated testing with doctest
One of the ways to make your code more robust to unexpected input is to develop with boundary cases in your mind. Test-driven code development begins with writing a set of unit tests for each class. These tests often includes normal and extreme use cases. Thanks to packages like doctest for Python, Mocha and Jasmine for Javascript etc., we can write and test codes with an easy format. In this blog post, I will present a short example of how to get started with doctest in Python. N.B. doctest is best suited for small tests with a few scripts. If you would like to run a system testing, look for some other packages!
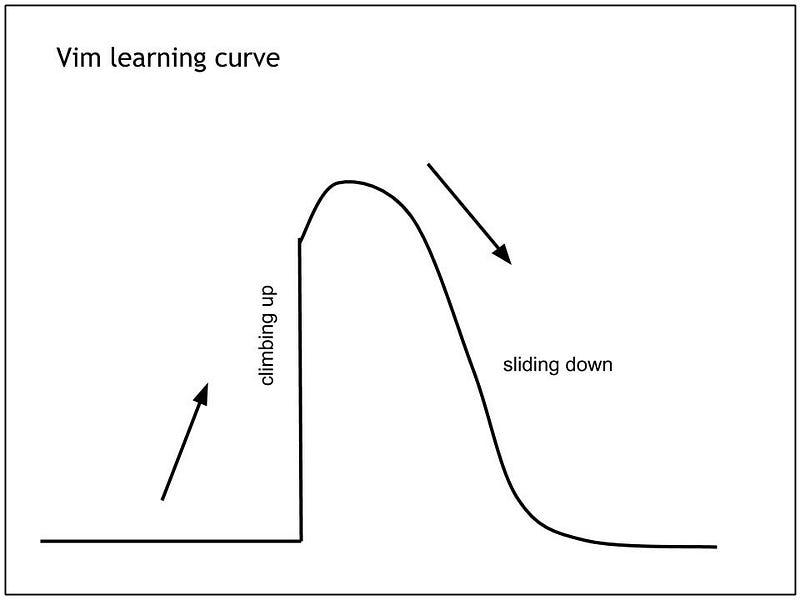
Vim and I
Vim is great. Despite its steep learning curve , it has many advantages and many loyal Vim followers will tell you that you should force yourself to use it.
{kind=link}
Personally I started using Vim when I was ssh-ing into the group servers or into my computer in department. In such scenarios, I could not open the IDEs with the nice GUIs 🙁 However, as time passed, Vim started to grow on me. Now, I can list a few reasons why I think it is great, for example, it requires a small amount of memory to run, has a short start up time and can handle large files pretty well.
Although, I am definitely not a Vim expert, I will tell you about some of the things I have added to my .vimrc. The .vimrc file is very handy for containing all your favourite settings, such as key mappings, custom commands, formatting and syntax highlighting. The file uses vimscript which is a programming language in itself. However, there is a lot of help online that tells you with what lines to add to your .vimrc. I would recommend installing Vundle which is a Vim plugin manager.
Here I will list some cool things that I have discovered you can do with your .vimrc. It has certainly made my life a bit nicer.
- Code Folding
Most IDEs provide a way to collapse functions and classes that results in only seeing the function/class definition and hiding the code. To do this in Vim add the following lines to your.vimrc" Enable folding set foldmethod=indent set foldlevel=99 " Enable folding with the spacebar nnoremap <space> za
Alternatively, you can install the Vim plugin SimpylFold. - Python indentation
Vim does not do auto indention like many IDEs. To automatically do PEP-8 indentation for Python, add the following to your .vimrc .
" PEP indentation au BufNewFile,BufRead *.py \ set tabstop=4 \ set softtabstop=4 \ set shiftwidth=4 \ set textwidth=79 \ set expandtab \ set autoindent \ set fileformat=unix
You can also install the Vim plugin vim-flake8 which is a static syntax and style checker for Python source code. It shows errors in a quickfix window and lets you jump to their location inside your code.
- Turn line numbers on
Rather than typing in:set nu
every time you open your files. You can always have them turned on by adding :set nu to your.vimrc - Autocompletion
When I switch from PyCharm to Vim I feel a bit lost without the autocompletion however, after a quick search I found many are using the Vim package Youcompleteme and it is awesome.
docopt for dummies
Parsing command line arguments is an annoying piece of boilerplate we all have to do. Documenting our code is either an absolutely essential part of software engineering, or a frivolous waste of research time, depending on who you ask. But what if I told you that we can combine the two? That you can handle your argument parsing simply by documenting how your code works? Well, the dream is now reality. Continue reading
Introduction to R Markdown
Two of our esteemed OPIGlets presented a workshop on collaborative research using Jupyter Notebook this week at ISMB in Chicago. Their workshop highlights the importance of finding ways to share your work conveniently and reproducibly. So on a related note, I thought I would share a brief introduction to another useful tool, R Markdown with RStudio, which I use to present updates to various supervisors and to remember what I did three months (or three days) ago. This method of sharing work is highly readable, reproducible, and narrative-driven.
I use R for much of my data analysis and all of my visualisation, and I count the tidyverse among my most beloved friends. If you’re so inclined, it’s easy to execute python, bash, and more from within R Markdown. You also don’t need to use RStudio to use R Markdown, but that’s a whole other story.
Starting a new markdown file in RStudio will generate a template script explaining most of what you need to know. If I showed you that then I’d be out of a blog post, but I will at least link to the R Markdown Reference Guide.
R Markdown files consist of text written in markdown, and code chunks that can be individually executed and displayed inline within RStudio. To “knit” the whole thing together, the knitr package is used to execute and combine code chunks, then pandoc converts the whole thing into an attractive document.
Here’s an example. The metadata at the top sets up the document. I’ll be generating an html document here, but notice some other tempting examples commented out. Yes, you can use it for Latex (swoon). You can even make a Word document, but really, why would you?
---
title: "Informative Title"
author: "Clare E. West"
date: "10/07/2018"
output: html_document
#output: beamer_presentation
#output: pdf_document
---
```{r setup, include=FALSE}
knitr::opts_chunk$set(echo = TRUE)
library(knitr)
library(ggplot2)
library(tidyr)
library(dplyr)
```
## Big Title
### Smaller title
R Markdown scripts have the extension .Rmd
R Markdown is __so__ *fun*. You can read all about it [here](https://www.rstudio.com/wp-content/uploads/2015/03/rmarkdown-reference.pdf).
```{r}
print("Hello world")
```
Notice that chunks are enclosed within three backticks, with the language and options in braces. Single commands can be executed inline using single backticks.
As highlighted in the example above, global options are set like this:
knitr::opts_chunk$set(echo = TRUE)
“echo=TRUE” means that the code in each chunk is displayed in the final product; this is useful to show collaborators (or your future self) exactly how you did something. Change this option (“echo = FALSE”) globally or in individual blocks to prevent code from printing. This is useful to hide uninteresting commands, or when presenting to people who don’t have the time or inclination to read your code (hard to imagine). Notice I’ve also used “include = FALSE” for the library-loading code chunk, which means evaluate but don’t include in the output. Another useful option is “eval = FALSE”, which means don’t even run this chunk.
So let’s see what that looks like when we render it:

The above example output as HTML

The above example output as Latex
Plots generated in code chunks or images from other sources can be embedded. Set the width in the options. “fig.width” sets the width (in inches) of the figure generated, while “out.width” scales the image in the final documents, for which the units will depend on the document type. Within RStudio, these are previewed inline below the code chunk.
## Including plots/images
```{r fig.width = 4, fig.height = 3, out.width = "400px", echo=FALSE}
t %>% group_by(Tour, Winner, N, Tournament) %>% filter(WRank <= 20) %>% summarise(WPts = max(WPts)) %>% ggplot(aes(x=N, y=WPts, group=Winner, colour=(Winner=="Murray A."))) + geom_point() + geom_line() + labs(x="Tournament Number",y="Ranking Points") + scale_colour_discrete("",labels=c("Not Andy Murray", "Andy Murray")) + theme_bw() + theme(legend.position = "bottom", legend.margin = margin(0, 0, 0, 0))
knitr::include_graphics("https://s.yimg.com/ny/api/res/1.2/69ZUzNSMYb09GKd8CNJeew--~A/YXBwaWQ9aGlnaGxhbmRlcjtzbT0xO3c9ODAwO2g9NjAw/http://media.zenfs.com/en_us/News/afp.com/0102e1f7d0d3c35303c8a62d56a5eb79c2c8b4d8.jpg")
```

Rather than just printing data R-style, you can nicely format it into a table using kable (part of knitr). I also style mine using kableExtra, which makes it look nice and gives you extra options. By default tables fill the full width, you can override this using e.g. kable_styling(full.width = FALSE, position = “left”). When making a latex document, use kable(table, booktabs = T, “latex”) to get a (reproducible) latex-style table.

Here’s how to use python and bash. Thanks to the package reticulate, you can even share objects between your R and Python chunks. Exclude reticulate (knitr::opts_chunk$set(python.reticulate=FALSE) if you prefer to keep your languages separate.
### Mix it up with python
```{python}
a='Wow python'
print(a.split()[0])
```
What a wild ride.
### or bash
```{bash, echo=TRUE}
ls | head
```
Oh look, there's our output, ready to share.

Finally, if you hate GUIs – and you know I do – you can ditch the interactive notebook part and just generate documents from R Markdown files like this:
rmarkdown::render("BlogExample.Rmd")
How to parse OAS data
We have recently released the Observed Antibody Space database – collection of cleaned and annotated antibody sequence (Ig-seq or AIRR-seq) data from 53 studies. We have formatted the data in the format that should facilitate data mining and since release we had several queries on how to parse the data out. Therefore here we give a small example of how to parse the data and make sense of it.
You should download the bulk data file from OAS, available here.
The datasets are separated into ‘data units‘ – collections of sequences that can be uniquely assigned to a range of metadata parameters such as study, organism etc. Our task therefore is to iterate through all those files and read sequences from each of these. Firstly we will attempt to iterate through the files and I will assume that you uncompressed the bulk data file into ../data/json folder. We will write a helper function that will simply list all files with its full paths in a directory and call it list_file_paths
import os
#Fetch all files in directory and subdirectories.
def list_file_paths(directory):
for dirpath,_,filenames in os.walk(directory):
for f in filenames:
yield os.path.abspath(os.path.join(dirpath, f))
if __name__ == '__main__':
#Replace this with the location of where you uncompressed the bulk data file.
directory = '../data/json'
for f in list_file_paths(directory):
print f
The code above will list all the files in ../data/json which incidentally are all the ‘data units’. Now our task is to parse out the output from each of the data units. They are gzipped files with data element on each line. Therefore we will use gzip library to stream the contents of a gzipped file rather than uncompressing each one of them separately. This is achieved by function parse_single_file
import os,gzip
#Fetch all files in directory and subdirectories.
def list_file_paths(directory):
for dirpath,_,filenames in os.walk(directory):
for f in filenames:
yield os.path.abspath(os.path.join(dirpath, f))
#Parse out the contents of a single file.
def parse_single_file(src):
#The first line are the meta entries.
meta_line = True
for line in gzip.open(src,'rb'):
print line
if __name__ == '__main__':
#Replace this with the location of where you uncompressed the bulk data file.
directory = '../data/json'
for f in list_file_paths(directory):
parse_single_file(f)
The code above will simply go through all the data unit files, stream the gzipped lines and print each one of them separately. Each line however is formatted as json – meaning it can be parsed using pythons json library and act as a pythonic dictionary. below we have parsed out the basic elements in the final incarnation of the code:
import os,gzip,json,pprint
#Fetch all files in directory and subdirectories.
def list_file_paths(directory):
for dirpath,_,filenames in os.walk(directory):
for f in filenames:
yield os.path.abspath(os.path.join(dirpath, f))
#Parse out the contents of a single file.
def parse_single_file(src):
#The first line are the meta entries.
meta_line = True
for line in gzip.open(src,'rb'):
if meta_line == True:
metadata = json.loads(line)
meta_line=False
print "Metadata:"
pprint.pprint(metadata)
continue
#Parse actual sequence data.
basic_data = json.loads(line)
print "Basic data:"
pprint.pprint(basic_data)
#IMGT-Numbered sequence.
print "IMGT-numbered sequence"
d = json.loads(basic_data['data'])
pprint.pprint(d)
print "==========="
if __name__ == '__main__':
#Replace this with the location of where you uncompressed the bulk data file.
directory = '../data/json'
for f in list_file_paths(directory):
parse_single_file(f)
The first line of each data unit are meta entries. These look as follows:
{u'Age': u'22-70',
u'Author': u'Halliley et al., (2015)',
u'BSource': u'Bone-Marrow',
u'BType': u'Plasma-B-Cells',
u'Chain': u'Heavy',
u'Disease': u'None',
u'Isotype': u'IGHM',
u'Link': u'https://doi.org/10.1016/j.immuni.2015.06.016',
u'Longitudinal': u'no',
u'Size': 934,
u'Species': u'human',
u'Subject': u'no',
u'Vaccine': u'Tetanus/Flu'}
The attributes should be self-explanatory and the existence of this data on top of each file is supposed to streamline searching through data-units if you wish to parse sequences given a particular configuration of meta-data entries (e.g. organism).
Next, the code parses out data on each sequence that is associated with its genes, full sequence, CDR3 and numbered sequence. Therefore the output for this will look something like this:
{u'cdr3': u'ARHQGVYWVTTAGLSH',
u'data': u'{"fwh1": {"11": "G", "24": "T", "13": "V", "12": "L", "15": "P", "14": "K", "17": "E", "16": "S", "19": "L", "18": "T", "22": "T", "26": "S", "25": "V", "21": "L", "20": "S", "23": "C"}, "fwh3": {"68": "N", "88": "S", "89": "L", "66": "Y", "67": "Y", "82": "T", "83": "S", "80": "V", "81": "D", "86": "Q", "87": "F", "84": "K", "85": "N", "92": "S", "79": "S", "69": "P", "104": "C", "78": "I", "77": "T", "76": "V", "75": "R", "74": "S", "72": "K", "71": "L", "70": "S", "102": "Y", "90": "K", "100": "A", "101": "V", "95": "T", "94": "V", "97": "A", "96": "A", "91": "L", "99": "T", "98": "D", "93": "S", "103": "Y"}, "fwh2": {"52": "W", "39": "W", "48": "Q", "49": "G", "46": "P", "47": "G", "44": "Q", "45": "P", "51": "E", "43": "R", "40": "G", "42": "I", "55": "S", "53": "I", "54": "G", "41": "W", "50": "L"}, "fwh4": {"120": "Q", "121": "G", "122": "T", "123": "L", "124": "V", "125": "P", "126": "V", "127": "S", "128": "S", "119": "G", "118": "W"}, "cdrh1": {"27": "G", "37": "Y", "31": "S", "30": "I", "28": "G", "29": "S", "35": "S", "34": "S", "38": "Y", "36": "S"}, "cdrh2": {"59": "S", "58": "Y", "57": "S", "56": "I", "63": "G", "64": "T", "65": "T"}, "cdrh3": {"111A": "W", "109": "G", "108": "Q", "115": "L", "114": "G", "117": "H", "116": "S", "111": "Y", "110": "V", "113": "A", "112": "T", "112A": "T", "112B": "V", "106": "R", "107": "H", "105": "A"}}',
u'j': u'IGHJ1*01',
u'name': 12,
u'redundancy': 1,
u'seq': u'GLVKPSETLSLTCTVSGGSISSSSYYWGWIRQPPGQGLEWIGSISYSGTTYYNPSLKSRVTISVDTSKNQFSLKLSSVTAADTAVYYCARHQGVYWVTTAGLSHWGQGTLVPVSS',
u'v': u'IGHV4-39*07'}
Above, redundancy refers to how many times we see a given sequence (seq) in a particular study. We also store the IMGT-numbered data (the data attribute) which needs a second round of json parsing and its output is a dictionary of IMGT-number – amino acid associations grouped by the regions of an antibody (cdrs and framework regions):
{u'cdrh1': {u'27': u'G',
u'28': u'G',
u'29': u'S',
u'30': u'I',
u'31': u'S',
u'34': u'S',
u'35': u'S',
u'36': u'S',
u'37': u'Y',
u'38': u'Y'},
u'cdrh2': {u'56': u'I',
u'57': u'S',
u'58': u'Y',
u'59': u'S',
u'63': u'G',
u'64': u'T',
u'65': u'T'},
u'cdrh3': {u'105': u'A',
u'106': u'R',
u'107': u'H',
u'108': u'Q',
u'109': u'G',
u'110': u'V',
u'111': u'Y',
u'111A': u'W',
u'112': u'T',
u'112A': u'T',
u'112B': u'V',
u'113': u'A',
u'114': u'G',
u'115': u'L',
u'116': u'S',
u'117': u'H'},
u'fwh1': {u'11': u'G',
u'12': u'L',
u'13': u'V',
u'14': u'K',
u'15': u'P',
u'16': u'S',
u'17': u'E',
u'18': u'T',
u'19': u'L',
u'20': u'S',
u'21': u'L',
u'22': u'T',
u'23': u'C',
u'24': u'T',
u'25': u'V',
u'26': u'S'},
u'fwh2': {u'39': u'W',
u'40': u'G',
u'41': u'W',
u'42': u'I',
u'43': u'R',
u'44': u'Q',
u'45': u'P',
u'46': u'P',
u'47': u'G',
u'48': u'Q',
u'49': u'G',
u'50': u'L',
u'51': u'E',
u'52': u'W',
u'53': u'I',
u'54': u'G',
u'55': u'S'},
u'fwh3': {u'100': u'A',
u'101': u'V',
u'102': u'Y',
u'103': u'Y',
u'104': u'C',
u'66': u'Y',
u'67': u'Y',
u'68': u'N',
u'69': u'P',
u'70': u'S',
u'71': u'L',
u'72': u'K',
u'74': u'S',
u'75': u'R',
u'76': u'V',
u'77': u'T',
u'78': u'I',
u'79': u'S',
u'80': u'V',
u'81': u'D',
u'82': u'T',
u'83': u'S',
u'84': u'K',
u'85': u'N',
u'86': u'Q',
u'87': u'F',
u'88': u'S',
u'89': u'L',
u'90': u'K',
u'91': u'L',
u'92': u'S',
u'93': u'S',
u'94': u'V',
u'95': u'T',
u'96': u'A',
u'97': u'A',
u'98': u'D',
u'99': u'T'},
u'fwh4': {u'118': u'W',
u'119': u'G',
u'120': u'Q',
u'121': u'G',
u'122': u'T',
u'123': u'L',
u'124': u'V',
u'125': u'P',
u'126': u'V',
u'127': u'S',
u'128': u'S'}}
We hope this quick intro to our data format will allow you to do great science with this data.