So, I recently deployed my first piece of scientific code. Well, sort of. I made a github with instructions on how to download, install and run it.
And then everyone broke it.
So, now having been on tech support duty for a few weeks, it seemed like a good idea to have a think about what I’ve learned.
Now, there is a big preface to this: the first and most important thing I learned is that I should do some reading on how to do this well. I have not yet done that reading, so this post isn’t so much going to offer any advice as catalogue my mistakes. Mistakes that will probably look extremely silly to anyone who has any familiarity with deployment, but might be interesting to anyone who doesn’t.
A surprising number of people really don’t want to touch the command line
Being a programmer who spends the vast majority of their time on the command line, invoking programs from there is very natural. As such, I very much underestimated the obstacle that even installing anaconda, a few packages, and cloning the source code would be. Even with instructions to copy and paste.
The issue is, if anything goes wrong, there is a good chance they don’t know whether it is my code or their environment breaking, which probably means they need to contact me about it (more on environments later).
Really, I probably could have saved myself an awful lot of support by making it an installable, and more with a gui to guide people through using the program.
Python is a pain
So, the first thing I learned was something I’d kind of been warned about: deploying python code is a pain in the butt. Especially to people who aren’t familiar with python, managing python environments is both tricky and overwhelming easy to break code with. Run a python script from the wrong environment and it is going to fail: if you are lucky with a failure to import a module, if you are unlucky with a cryptic error due to say changes between various python versions.
Speaking of python versions, developing in 3.9 and not testing in 3.7 then telling people to install that can result in a surprising number of surprisingly difficult bugs.
The instructions weren’t clear enough
Scientific code I think generally caters an awful lot to expert users, people who really understand the model and even are willing to open the source code to figure out the implementation.
My first stab at documentation managed to not be clear enough to the people who didn’t want to touch the command line and those who were willing to open the source code because they wanted to do something spicy.
So yeah, good documentation is an acquired skill.
Distributed computing is a nightmare
In principle, distribution is terrific: get a library that will allow you to reduce running arbitrary python code on multiple nodes to a simple map-like interface. On big clusters, like a lot of scientists use, this can mean speed ups from 10 to even 1000 times.
The only problem is, everyone’s cluster is a special snowflake, and you can’t access most of them to fix things. This can make iteration with a non-programmer painfully slow.
Libraries don’t help as much as I’d have thought either: indeed, my experience of Dask and Dask Jobqueue has been a consistently uphill battle. From the fact that my workload likes individual nodes sharing lots of memory and a few cpus to some truly arcane errors (one that broke in the msgpack code), I have generally considered (and even started) writing my own code to do this.
Active development doesn’t reach people
Code that is being updated several times a day in response to bugfixes can be great – but if people aren’t pulling and installing it, no-one is going to benefit. I’m seriously tempted to write some code to either auto-update on running or at least let folk know it has been updated.
Summary
In summary, a lot went wrong in my first stab at this. Very much come to appreciate a good deployment is an artform, and I’ve got an awful lot of reading to do. In particular, the above problem areas really have eaten a lot of time that probably could have been used doing actual science with the code, so there is a good incentive to get it right.




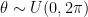 . Extending this idea to 3D rotations, we could sample each of the three Euler angles from the same uniform distribution
. Extending this idea to 3D rotations, we could sample each of the three Euler angles from the same uniform distribution 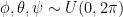 . This, however, gives more probability density to transformations which are clustered towards the poles:
. This, however, gives more probability density to transformations which are clustered towards the poles:
 into two sets – a training set
into two sets – a training set  and a test set
and a test set 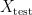 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 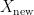 which it will encounter in the future. Right?
which it will encounter in the future. Right?