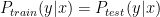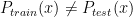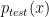In early July I attended the the Seventh Joint Sheffield Conference on Cheminformatics. There was a variety of talks with speakers at all stages of their career. I was lucky enough to be invited to speak at the conference, and gave my first conference talk! I have written two blog posts about the conference: part 1 briefly describes a talk that I found interesting and part 2 describes the work I spoke about at the conference.
One of the most interesting parts of the conference was the active twitter presence. #ShefChem16. All of the talks were live tweeted which provided a summary of each talk and also included links to software or references. It also allowed speakers to gain insight and feedback on their talk instantly.
One of the talks I found most interesting presented the Protein-Ligand Interaction Profiler (PLIP). It is a method for the detection of protein-ligand interactions. PLIP is open-source and has a web-based online tool and a command-line tool. Unlike PyMol which only calculates polar contacts, and not the type of interaction, PLIP calculates 8 different types of interactions: hydrogen bonding, hydrophobic, π-π stacking, π-cation interactions, salt bridges, water bridges, halogen bonds, metal complexes. For a given pdb file the interactions are calculated and shown in a publication quality figure shown here.

The display can also be downloaded as a PyMol session so the display can be modified.
This tool is an extremely useful way to calculate protein-ligand interactions and can be used to find the types of interactions formed by the protein-ligand complex.
PLIP can be found here: https://projects.biotec.tu-dresden.de/plip-web/plip/