
Now that PDBbind 2020 has been released, I want to draw some attention to an issue with using the SDF files that are supplied in the PDBbind refined set 2020.
Normally, SDF files save the chirality information of compounds in the atom block of the file which is shown belowas a snipped of the full sdf file for the ligand of PDB entry 4qsv. The column that defines chirality is marked in red.

As you can see, all columns shown here are 0. The SDF files supplied by PDBbind for some reason do NOT encode chirality information explicitly. This will be a problem when using RDKit to read the molecule and transform it into a smiles string. By using the following commands to read the ligand for 4qsv from PDBBind 2020 and write a SMILES string, we get:
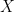 into two sets – a training set
into two sets – a training set 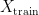 and a test set
and a test set 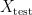 . The model is then subsequently trained on the examples in the training set
. The model is then subsequently trained on the examples in the training set 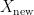 which it will encounter in the future. Right?
which it will encounter in the future. Right?

.jpg){kind=link}