
In this blog post I will share with you the materials that I found most useful when I started doing some Bayesian Optimisation in my research. Bear in mind, I am a Chemist by training, so I approached this topic from a non-mathematical background (my eyes have to be persuaded to look at mathematical equations). Out of all the materials I have come across, I found these to be the most accessible.
Continue readingAuthor Archives: Susan Leung
Trying out some code from the Eighth Joint Sheffield Conference on Chemoinformatics: finding the most common functional groups present in the DSPL library
Last month a bunch of us attended the Sheffield Chemoinformatics Conference. We heard many great presentations and there were many invitations to check out one’s GitHub page. I decided now is the perfect time to try out some code that was shown by one presenter.
Peter Ertl from Novartis presented his work on the The encyclopedia of functional groups. He presented a method that automatically detects functional groups, without the use of a pre-defined list (which is what most other methods use for detecting functional groups). His method involves recursive searching through the molecule to identify groups of atoms that meet certain criteria. He used his method to answer questions such as: how many functional groups are there and what are the most common functional groups found in common synthetic molecules versus bioactive molecules versus natural products. Since I, like many others in the group, are interested in fragment libraries (possibly due to a supervisor in common), I thought I could try it out on one of these.
Continue readingSpring 2019 ACS National Meeting (Orlando)
This blog post is jointly written by Lucian, Joe and Susan who recently attended the Spring ACS National Meeting 2019.

The Spring ACS National Meeting was held in sunny Orlando, Florida and was a five day event (29th March – 4th April). The temperature averaged 25°C , which was amazing compared to the UK (sorry) and meant we all got a lovely tan. We all presented our work in the form of talks in the divisions of COMP or CINF but in this blog post we write about our highlights of the conference.
Vim and I
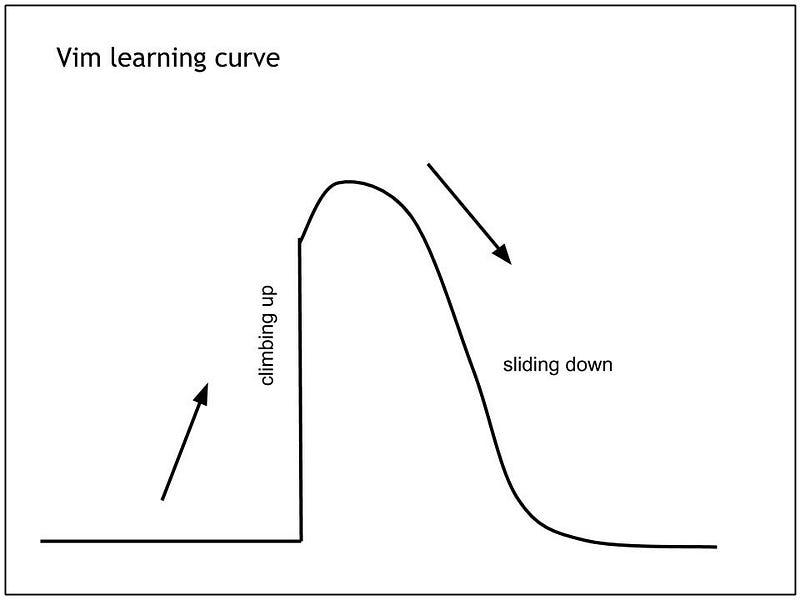
Vim is great. Despite its steep learning curve , it has many advantages and many loyal Vim followers will tell you that you should force yourself to use it.
{kind=link}
Personally I started using Vim when I was ssh-ing into the group servers or into my computer in department. In such scenarios, I could not open the IDEs with the nice GUIs 🙁 However, as time passed, Vim started to grow on me. Now, I can list a few reasons why I think it is great, for example, it requires a small amount of memory to run, has a short start up time and can handle large files pretty well.
Although, I am definitely not a Vim expert, I will tell you about some of the things I have added to my .vimrc. The .vimrc file is very handy for containing all your favourite settings, such as key mappings, custom commands, formatting and syntax highlighting. The file uses vimscript which is a programming language in itself. However, there is a lot of help online that tells you with what lines to add to your .vimrc. I would recommend installing Vundle which is a Vim plugin manager.
Here I will list some cool things that I have discovered you can do with your .vimrc. It has certainly made my life a bit nicer.
- Code Folding
Most IDEs provide a way to collapse functions and classes that results in only seeing the function/class definition and hiding the code. To do this in Vim add the following lines to your.vimrc" Enable folding set foldmethod=indent set foldlevel=99 " Enable folding with the spacebar nnoremap <space> za
Alternatively, you can install the Vim plugin SimpylFold. - Python indentation
Vim does not do auto indention like many IDEs. To automatically do PEP-8 indentation for Python, add the following to your .vimrc .
" PEP indentation au BufNewFile,BufRead *.py \ set tabstop=4 \ set softtabstop=4 \ set shiftwidth=4 \ set textwidth=79 \ set expandtab \ set autoindent \ set fileformat=unix
You can also install the Vim plugin vim-flake8 which is a static syntax and style checker for Python source code. It shows errors in a quickfix window and lets you jump to their location inside your code.
- Turn line numbers on
Rather than typing in:set nu
every time you open your files. You can always have them turned on by adding :set nu to your.vimrc - Autocompletion
When I switch from PyCharm to Vim I feel a bit lost without the autocompletion however, after a quick search I found many are using the Vim package Youcompleteme and it is awesome.
My experience with (semi-)automating organic synthesis in the lab
After three years of not touching a single bit of glassware, I have recently donned on the white coat and stepped back into the Chemistry lab. I am doing this for my PhD project to make some of the follow-up compounds that my pipeline suggests. However, this time there is a slight difference – I am doing reactions with the aid of a liquid handler robot, the Opentrons. This is the first encounter that I have with (semi-)automated synthesis and definitely a very exciting opportunity! (Thanks to my industrial sponsor, Diamond Light Source!)

A picture of the Opentrons machine I have been using to do some organic reactions. Picture taken from https://opentrons.com/robots.
Opentrons is primarily used by biologists and their goal is to make a platform to easily share protocols and reproduce each other’s work (I think we can all agree how nice this would be!). They provide a very easy to use API, wishing it to be accessible to any bench scientist with basic computer skills. From my experience so far, this has been the case as I found it extremely easy to pick up and write my own protocols for chemical reactions. Here is the command that will: (1) pick up a new pipette tip; (2) transfer a volume from source1 to destination1; (3) drop the pipette tip in the trash; (4) pick up a new pipette tip; (5) transfer a volume from source2 to destination2; (5) drop the pipette tip in the trash.
pipette.transfer(volume, [source1, source2], [destination1, destination2], new_tip=’always')
But of course not everything is plain sailing – there are many challenges you will encounter by using an automated pipette. The robot is a liquid handler – it cannot handle solids so either the solids need to be pre-weighed and/or made into solution beforehand. Further difficulties lie within the properties of the solvent it is handling, for example:
- Dripping – low boiling point solvents tend to drip more.
- Viscosity of liquids causes issues with not drawing up the correct amount of liquid – more viscous liquids require longer times to aspirate and if aspiration is too quick then air pockets may be drawn up.
Here is a GIF I made of a dry run I was doing with the robot (sorry for the slight shake, this was recorded on my phone in the lab… See their website for professional footage of the robot!)

My (shaky) footage of a dry run I was performing with the Opentrons.
When Does Chemical Elaboration Induce a Ligand To Change Its Binding Mode?
When Does Chemical Elaboration Induce a Ligand To Change Its Binding Mode?
For my journal club in June, I chose to present a Journal of Medicinal Chemistry article entitled “When Does Chemical Elaboration Induce a Ligand To Change Its Binding Mode?” by Malhotra and Karanicolas. This article uses a large scale collection of ligand pairs to investigate the circumstances in which elaborations of a ligand change the original binding mode.
One of the primary goals in medicinal chemistry is the optimisation of biological activity by chemical elaboration of a hit compound. This hit-to-lead optimisation often assumes that addition of functional groups to a given hit scaffold will not change the original binding mode.
In order to investigate the circumstances in which this assumption holds true and how often it holds true, they built up a large-scale collection of 297 related ligand pairs solved in complex with the same protein partner. Each pair consisted of a larger and smaller ligand; the larger ligand could have arisen from elaboration of the smaller ligand. They found that for 41 out of the 297 pairs (14%), the binding mode changed upon elaboration of the smaller ligand.
They investigated many physicochemical properties of the ligand, the protein-ligand complex and the protein binding pocket. They summarise the statistical significance and predictive power of the investigated properties with the table shown below.

They found that the property with the lowest p-value was the “rmsd after minimisation of the aligned complex” (RMAC). They developed this metric to probe whether the larger ligand could be accommodated in the protein without changing binding mode. They did so by aligning the shared substructure of the larger ligand onto the smaller ligand’s complex and then carrying out an energy minimisation. By monitoring the RMSD difference of the larger ligand relative to the initial pose (RMAC), they can gauge how compatible the larger ligand is with the protein. Larger RMAC values indicate greater incompatibility, hence a greater likelihood for the binding mode to not be preserved.
The authors generated receiver operating characteristic (ROC) plots to compare the predictive power of the properties considered. ROC curves are made by plotting the true positive rate (TPR) against the false positive rate (FPR). A random classifier would yield the dotted line from the bottom left to the top right, shown in the plots below. The best predictors would give a point in the top left corner of the plot. The properties that do well include RMAC, pocket volume, molecular weight, lipophilicity and potency.
 They also combined properties to enhance predictive power and conclude that RMAC and molecular weight together offers good predictivity.
They also combined properties to enhance predictive power and conclude that RMAC and molecular weight together offers good predictivity. Finally, the authors look at the pairs that have low RMAC values (i.e. the elaboration should be compatible with the protein pocket), yet show a change in binding mode. For these cases, a specific substitution may enable formation of a new, stronger interaction or for pseudosymmetric ligands, the alternate pose can mimic many of the interactions of the original pose.
Finally, the authors look at the pairs that have low RMAC values (i.e. the elaboration should be compatible with the protein pocket), yet show a change in binding mode. For these cases, a specific substitution may enable formation of a new, stronger interaction or for pseudosymmetric ligands, the alternate pose can mimic many of the interactions of the original pose.
Using RDKit to load ligand SDFs into Pandas DataFrames
If you have downloaded lots of ligand SDF files from the PDB, then a good way of viewing/comparing all their properties would be to load it into a Pandas DataFrame.
RDKit has a very handy function just for this – it’s found under the PandasTool module.
I show an example below within Jupypter-notebook, in which I load in the SDF file, view the table of molecules and perform other RDKit functions to the molecules.
First import the PandasTools module:
from rdkit.Chem import PandasTools
Read in the SDF file:
SDFFile = "./Ligands_noHydrogens_noMissing_59_Instances.sdf" BRDLigs = PandasTools.LoadSDF(SDFFile)
You can see the whole table by calling the dataframe:
BRDLigs

The ligand properties in the SDF file are stored as columns. You can view what these properties are, and in my case I have loaded 59 ligands each having up to 26 properties:
BRDLigs.info()

It is also very easy to perform other RDKit functions on the dataframe. For instance, I noticed there is no heavy atom column, so I added my own called ‘NumHeavyAtoms’:
BRDLigs['NumHeavyAtoms']=BRDLigs.apply(lambda x: x['ROMol'].GetNumHeavyAtoms(), axis=1)
Here is the column added to the table, alongside columns containing the molecules’ SMILES and RDKit molecule:
BRDLigs[['NumHeavyAtoms','SMILES','ROMol']]
