
Introduction
An epidemic is sweeping through cheminformatics (and machine learning) research: ugly results tables. These tables are typically bloated with metrics (such as regression and classification metrics next to each other), vastly differing tasks, erratic bold text, and many models. As a consequence, results become difficult to analyse and interpret. Additionally, it is rare to see convincing evidence, such as statistical tests, for whether one model is ‘better’ than another (something Pat Walters has previously discussed). Tables are a practical way to present results and are appropriate in many cases; however, this practicality should not come at the cost of clarity.
The terror of ugly tables extends to benchmark leaderboards, such as Therapeutic Data Commons (TDC). These leaderboard tables do not show:
- whether differences in metrics between methods are statistically significant,
- whether methods use ensembles or single models,
- whether methods use classical (such as Morgan fingerprints) or learned (such as Graph Neural Networks) representations,
- whether methods are pre-trained or not,
- whether pre-trained models are supervised, self-supervised, or both,
- the data and tasks that pre-trained models are pre-trained on.
This lack of context makes meaningful comparisons between approaches challenging, obscuring whether performance discrepancies are due to variance, ensembling, overfitting, exposure to more data, or novelties in model architecture and molecular featurisation. Confirming the statistical significance of performance differences (under consistent experimental conditions!) is crucial in constructing a more lucid picture of machine learning in drug discovery. Using figures to share results in a clear, non-tabular format would also help.
Statistical validation is particularly relevant in domains with small datasets, such as drug discovery, as the small number of test samples leads to high variance in performance between different splits. Recent work by Ash et al. (2024) sought to alleviate the lack of statistical validation in cheminformatics by sharing a helpful set of guidelines for researchers. Here, we explore implementing some of the methods they suggest (plus some others) in Python.
Imports
Imports
import lightgbm as lgb
import matplotlib.pyplot as plt
from matplotlib.colors import ListedColormap
import numpy as np
import seaborn as sns
import pandas as pd
import polaris as po
from rdkit import Chem
from rdkit.Chem import Descriptors
from rdkit.DataStructs import TanimotoSimilarity
from rdkit.ML.Cluster import Butina
from rdkit.ML.Descriptors import MoleculeDescriptors
from rdkit.Chem.rdFingerprintGenerator import GetMorganGenerator, GetMorganFeatureAtomInvGen
from scipy import stats
from sklearn.metrics import mean_absolute_error, r2_score
from tqdm import tqdm
from sklearn.model_selection import GroupKFold
Below are the various packages, and their versions, used for this blog.
| Module | Version |
|---|---|
| lightgbm | 4.5.0 |
| matplotlib | 3.9.2 |
| numpy | 1.26.4 |
| pandas | 2.2.3 |
| polaris | 0.8.6 |
| rdkit | 2024.03.5 |
| scipy | 1.14.1 |
| seaborn | 0.13.2 |
| sklearn | 1.6.0 |
| tqdm | 4.66.5 |
Dataset
The first thing we need is a benchmark dataset. Luckily, Polaris has numerous benchmarking datasets for drug discovery-related tasks. First, we need to log in to Polaris (which you can do with a GitHub account), and then we need to choose a dataset. If you do not want to log in to Polaris, the original CSV for the dataset used here can be found in this repository.
I opted to use a dataset from Fang et al. (2023) as it fulfills several criteria:
- the labels are all drug discovery-related molecular property prediction tasks,
- the data is non-federated, i.e., there is only one source (which should reduce noise),
- some benchmarks have very few examples, as is typical in drug discovery.
Polaris login
po.hub.client.PolarisHubClient().login()Display dataset
df = dataset.table
df = df.drop('UNIQUE_ID', axis=1)
df.head()| MOL_SMILES | SMILES | LOG_HLM_CLint | LOG_RLM_CLint | LOG_MDR1-MDCK_ER | LOG_HPPB | LOG_RPPB | LOG_SOLUBILITY | |
|---|---|---|---|---|---|---|---|---|
| 0 | Brc1cc… | Brc1cc… | 0.886265 | 2.357933 | -0.247518 | NaN | NaN | 1.536432 |
| 1 | Brc1cc… | Brc1cc… | 0.675687 | 1.613704 | -0.010669 | NaN | NaN | 1.797475 |
| 2 | Brc1cn… | Brc1cn… | 2.081607 | 3.753651 | NaN | NaN | NaN | NaN |
| 3 | Brc1cn… | Brc1cn… | NaN | NaN | NaN | NaN | NaN | -0.033858 |
| 4 | Brc1nn… | Brc1nn… | 1.888410 | 3.492201 | -0.235024 | NaN | NaN | NaN |
The MOL_smiles column contains the molecular SMILES strings after standardisation. The SMILES column contains the original molecular SMILES strings.
There are six endpoints in this dataset from in vitro experimental assays:
- LOG_HLM_CLint; a measure of drug clearance by human liver cells,
- LOG_RLM_CLint; a measure of drug clearance by rat liver cells
- LOG_MDR1-MDCK_ER; a measure of active transport by overexpressed P-glycoprotein 1 (aka multidrug resistance protein 1 [MDR1]) in Madin-Darby canine kidney cells (MDCK) cells,
- LOG_HPPB; a measure of plasma protein binding in humans,
- LOG_RPPB; a measure of plasma protein binding in rats,
- LOG_SOLUBILITY; a measure of aqueous solubility.
In total there are 3521 molecules in this dataset:
Dataset size
f'Dataset size: {df.shape}''Dataset size: (3521, 9)'However, not every molecule has a value for every endpoint. Let’s take a look at human plasma protein binding:
Select task
df = df[['MOL_smiles', 'LOG_HPPB']]
df = df.dropna()
f'Human plasma protein binding dataset size: {df.shape}''Human plasma protein binding dataset size: (194, 2)'There are only 194 molecules in this benchmark, meaning performance variance will likely be high between different test splits.
Featurisation
Now that we have a dataset of molecules and target labels, we need to featurise the molecules. First, let’s convert the SMILES strings to RDKit molecules and extract the target labels:
Convert SMILES
mols = [Chem.MolFromSmiles(i) for i in df.MOL_smiles]
y_true = df.LOG_HPPB.to_numpy()
Three commonly used molecular featurisation methods are physicochemical descriptor vectors (PDV), extended-connectivity fingerprints (ECFP), and functional class fingerprints (FCFP). PDVs are vectors containing numerical molecular features, such as molecular weight and total charge. ECFPs are bit vector representations of 2D molecular topology, where 1 indicates the presence of a substructure, and 0 indicates the absence of a substructure. FCFPs are a variant of ECFPs that group atoms by certain features (e.g., all halogen atoms are labelled the same). See Rogers and Hahn (2010) and McGibbon et al. (2024) for more.
First, lets generate PDVs for each molecule with RDKit:
PDV descriptors list
# List of RDKit molecular descriptors
descriptor_list = [
'BalabanJ', 'BertzCT', 'Chi0', 'Chi0n', 'Chi0v', 'Chi1', 'Chi1n', 'Chi1v', 'Chi2n', 'Chi2v',
'Chi3n', 'Chi3v', 'Chi4n', 'Chi4v', 'EState_VSA1', 'EState_VSA10', 'EState_VSA11',
'EState_VSA2', 'EState_VSA3', 'EState_VSA4', 'EState_VSA5', 'EState_VSA6', 'EState_VSA7',
'EState_VSA8', 'EState_VSA9', 'ExactMolWt', 'FractionCSP3', 'HallKierAlpha', 'HeavyAtomCount', 'HeavyAtomMolWt',
'Ipc', 'Kappa1', 'Kappa2', 'Kappa3', 'LabuteASA', 'MaxAbsEStateIndex', 'MaxAbsPartialCharge',
'MaxEStateIndex', 'MaxPartialCharge', 'MinAbsEStateIndex', 'MinAbsPartialCharge',
'MinEStateIndex', 'MinPartialCharge', 'MolLogP', 'MolMR', 'MolWt', 'NHOHCount', 'NOCount',
'NumAliphaticCarbocycles', 'NumAliphaticHeterocycles', 'NumAliphaticRings',
'NumAromaticCarbocycles', 'NumAromaticHeterocycles', 'NumAromaticRings', 'NumHAcceptors',
'NumHDonors', 'NumHeteroatoms', 'NumRadicalElectrons', 'NumRotatableBonds',
'NumSaturatedCarbocycles', 'NumSaturatedHeterocycles', 'NumSaturatedRings',
'NumValenceElectrons', 'PEOE_VSA1', 'PEOE_VSA10', 'PEOE_VSA11', 'PEOE_VSA12', 'PEOE_VSA13',
'PEOE_VSA14', 'PEOE_VSA2', 'PEOE_VSA3', 'PEOE_VSA4', 'PEOE_VSA5', 'PEOE_VSA6', 'PEOE_VSA7',
'PEOE_VSA8', 'PEOE_VSA9', 'RingCount', 'SMR_VSA1', 'SMR_VSA10', 'SMR_VSA2', 'SMR_VSA3',
'SMR_VSA4', 'SMR_VSA5', 'SMR_VSA6', 'SMR_VSA7', 'SMR_VSA8', 'SMR_VSA9', 'SlogP_VSA1',
'SlogP_VSA10', 'SlogP_VSA11', 'SlogP_VSA12', 'SlogP_VSA2', 'SlogP_VSA3', 'SlogP_VSA4',
'SlogP_VSA5', 'SlogP_VSA6', 'SlogP_VSA7', 'SlogP_VSA8', 'SlogP_VSA9', 'TPSA', 'VSA_EState1',
'VSA_EState10', 'VSA_EState2', 'VSA_EState3', 'VSA_EState4', 'VSA_EState5', 'VSA_EState6',
'VSA_EState7', 'VSA_EState8', 'VSA_EState9', 'fr_Al_COO', 'fr_Al_OH', 'fr_Al_OH_noTert',
'fr_ArN', 'fr_Ar_COO', 'fr_Ar_N', 'fr_Ar_NH', 'fr_Ar_OH', 'fr_COO', 'fr_COO2', 'fr_C_O',
'fr_C_O_noCOO', 'fr_C_S', 'fr_HOCCN', 'fr_Imine', 'fr_NH0', 'fr_NH1', 'fr_NH2', 'fr_N_O',
'fr_Ndealkylation1', 'fr_Ndealkylation2', 'fr_Nhpyrrole', 'fr_SH', 'fr_aldehyde',
'fr_alkyl_carbamate', 'fr_alkyl_halide', 'fr_allylic_oxid', 'fr_amide', 'fr_amidine',
'fr_aniline', 'fr_aryl_methyl', 'fr_azide', 'fr_azo', 'fr_barbitur', 'fr_benzene',
'fr_benzodiazepine', 'fr_bicyclic', 'fr_diazo', 'fr_dihydropyridine', 'fr_epoxide',
'fr_ester', 'fr_ether', 'fr_furan', 'fr_guanido', 'fr_halogen', 'fr_hdrzine',
'fr_hdrzone', 'fr_imidazole', 'fr_imide', 'fr_isocyan', 'fr_isothiocyan', 'fr_ketone',
'fr_ketone_Topliss', 'fr_lactam', 'fr_lactone', 'fr_methoxy', 'fr_morpholine', 'fr_nitrile',
'fr_nitro', 'fr_nitro_arom', 'fr_nitro_arom_nonortho', 'fr_nitroso', 'fr_oxazole',
'fr_oxime', 'fr_para_hydroxylation', 'fr_phenol', 'fr_phenol_noOrthoHbond', 'fr_phos_acid',
'fr_phos_ester', 'fr_piperdine', 'fr_piperzine', 'fr_priamide', 'fr_prisulfonamd',
'fr_pyridine', 'fr_quatN', 'fr_sulfide', 'fr_sulfonamd', 'fr_sulfone', 'fr_term_acetylene',
'fr_tetrazole', 'fr_thiazole', 'fr_thiocyan', 'fr_thiophene', 'fr_unbrch_alkane', 'fr_urea', 'qed'
]PDV generation
# Physicochemical descriptor vector generation
desc_gen = MoleculeDescriptors.MolecularDescriptorCalculator(descriptor_list)
pdvs = np.array([desc_gen.CalcDescriptors(m) for m in mols])Next, let’s generate ECFP fingerprints with a maximum substructure radius of 2 and bit length of 1024 (ECFP4
ECFP generation
# Morgan fingerprint generation
ecfp_gen = GetMorganGenerator(radius=2, fpSize=1024, includeChirality=True)
fps = np.array([ecfp_gen.GetFingerprintAsNumPy(m) for m in mols])Now, let’s generate FCFP fingerprints with a maximum substructure radius of 2 and bit length of 1024 (FCFP4
FCFP generation
# Morgan functional fingerprint generation
fcfp_gen = GetMorganGenerator(radius=2, fpSize=1024, includeChirality=True, atomInvariantsGenerator=GetMorganFeatureAtomInvGen())
func_fps = np.array([fcfp_gen.GetFingerprintAsNumPy(m) for m in mols])
Due to the small number of dataset samples, we should apply feature selection and dimensionality reduction to each featurisation method on training set data. However, for brevity, this step will be excluded.
Splitting
Now, we need to work out how to split our data. To compare generalisability between methods, we need to minimise data leakage between training and testing splits; this is achievable through implementing a covariate shift, i.e., ensuring the training set and testing set molecules are dissimilar. There are several methods for splitting molecular data into hypothetically dissimilar subsets, including scaffold splitting, Butina clustering, and spectral clustering. Here, we look at Butina clustering, which is implementable in RDKit.
First, we must calculate the pairwise Tanimoto similarity (aka Jaccard index) over our benchmark molecules. Tanimoto similarity is a method for calculating the bit overlap between binary vectors and is commonly applied to ECFP fingerprints to measure similarity between molecules. The maximum value of Tanimoto similarity is 1, indicating the bit vectors are identical, and the minimum value is 0, indicating the bit vectors possess no common on-bits. Pairwise calculation of Tanimoto similarity for a small number of molecules is easily implementable in Python with NumPy:
Tanimoto similarity
def tanimoto_pairwise(fps: np.ndarray) -> np.ndarray:
"""
Compute the pairwise Tanimoto similarity for a set of fingerprints.
Parameters:
fps (np.ndarray)
A 2D numpy array where each row represents a binary fingerprint.
Returns:
np.ndarray
A 2D numpy array containing the Tanimoto similarity coefficients
for each pair of fingerprints. The element at position (i, j) in
the output array represents the Tanimoto similarity coefficient
between the i-th and j-th fingerprints in the input array.
See:
https://doi.org/10.1021/ci300261r
https://doi.org/10.1111/j.1469-8137.1912.tb05611.x
"""
c = np.matmul(fps, fps.T)
ab = np.diag(c)
ab = np.add.outer(ab, ab)
return c / (ab - c)
We can cluster the benchmark molecules based on Tanimoto similarity between molecules. Here, Butina.ClusterData takes as input the flattened lower triangle of the similarity distance matrix, a threshold distance value for whether two molecules are considered neighbours, a boolean to show that the input data is distance values, and the number of molecules to cluster:
Butina clustering
# get pairwise distances based on Tanimoto similarity
dist_matrix = 1 - tanimoto_pairwise(fps)
# flatten lower triangle of symmetric matrix
dist_data = dist_matrix[np.tril_indices(len(dist_matrix), -1)]
# butina clustering
clusters = Butina.ClusterData(
dist_data, distThresh=0.65, isDistData=True, nPts=len(dist_matrix)
)
# map molecules to clusters in an array
butina_groups = np.zeros(len(fps))
for i in range(len(clusters)):
for j in clusters[i]:
butina_groups[j] = i
f'Number of clusters: {len(clusters)}''Number of clusters: 126'We have molecules clustered by topological similarity. Now, we need a method for splitting our data given the clusters. Enter GroupKFold. Given a set of groups, GroupKFold produces K-Fold cross-validation train and test sets where splits are based on groups (i.e., clusters) rather than individual data points:

We can use GroupKFold to split our molecular data based on Butina clusters. This approach can also be applied to scaffold groupings and spectral clusters. Additionally, random seeding and group shuffling were added to GroupKFold in version 1.6 of sklearn, so cross-validation can be repeated to generate many different train-test splits. For example:
GroupKFold
print(f'Butina clusters for first 10 molecules: {butina_groups[:10]}')
for repeat in range(2):
folds = 2
splitter = GroupKFold(n_splits=folds, shuffle=True, random_state=repeat)
print(f'Repeatition {repeat}:')
for num, fold in enumerate(splitter.split(fps[:10], groups=butina_groups[:10])):
print(f'Fold {num}:')
train_idx, test_idx = fold
train_groups, test_groups = [], []
for i in train_idx:
train_groups.append(butina_groups[i])
for i in test_idx:
test_groups.append(butina_groups[i])
print(f'Train groups: {np.unique(train_groups)}')
print(f'Test groups: {np.unique(test_groups)}')Butina clusters for first 10 molecules: [125 124 0 0 0 0 29 9 9 9]
Repeatition 0:
Fold 0:
Train groups: [124 125]
Test groups: [ 0 9 29]
Fold 1:
Train groups: [ 0 9 29]
Test groups: [124 125]
Repeatition 1:
Fold 0:
Train groups: [ 0 124]
Test groups: [ 9 29 125]
Fold 1:
Train groups: [ 9 29 125]
Test groups: [ 0 124]Therefore, we can generate many cross-validation splits with dissimilar train and test molecules.
Training and Testing
Finally, we need to train and test models on each of our molecular representations over our Butina splits. Here, I’m using light gradient boosted machines from lightgbm (docs and paper) due to their fast training and inference. As this is a regression task, we can use LGBMRegressor.
For recording performance, we can use two different regresseion metrics; mean absolute error (MAE), and the coefficient of determination (
Run training and testing
# set number of repeats and cross-validation folds
reps = 100
num_folds = 5
total_splits = reps*num_folds
# output results storage
results = {
'ecfps': {
'mae': np.zeros(total_splits),
'r2': np.zeros(total_splits),
},
'func_fps': {
'mae': np.zeros(total_splits),
'r2': np.zeros(total_splits),
},
'pdv': {
'mae': np.zeros(total_splits),
'r2': np.zeros(total_splits),
}
}
counter = 0
pbar = tqdm(total=total_splits)
for i in range(reps):
# loop over repetitions
# initiate splitter with new random state
splitter = GroupKFold(n_splits=num_folds, shuffle=True, random_state=i)
# split into folds based on butina clusters
for fold in splitter.split(fps, groups=butina_groups):
# loop over folds
train_idx, test_idx = fold
# get train and test y values
y_train = y_true[train_idx]
y_test = y_true[test_idx]
# fit LGBM model and predict using Morgan Fingerprints
X_train = fps[train_idx]
X_test = fps[test_idx]
model = lgb.LGBMRegressor(verbose=-1)
model.fit(X_train, y_train)
preds = model.predict(X_test)
results['ecfps']['mae'][counter] = mean_absolute_error(y_test, preds)
results['ecfps']['r2'][counter] = r2_score(y_test, preds)
# fit LGBM model and predict using functional Morgan Fingerprints
X_train = func_fps[train_idx]
X_test = func_fps[test_idx]
model = lgb.LGBMRegressor(verbose=-1)
model.fit(X_train, y_train)
preds = model.predict(X_test)
results['func_fps']['mae'][counter] = mean_absolute_error(y_test, preds)
results['func_fps']['r2'][counter] = r2_score(y_test, preds)
# fit LGBM model and predict using physicochemical descriptor vectors
X_train = pdvs[train_idx]
X_test = pdvs[test_idx]
model = lgb.LGBMRegressor(verbose=-1)
model.fit(X_train, y_train)
preds = model.predict(X_test)
results['pdv']['mae'][counter] = mean_absolute_error(y_test, preds)
results['pdv']['r2'][counter] = r2_score(y_test, preds)
counter += 1
pbar.update(1)
pbar.close()100%|████████████████████████████████████████████████████████████████████| 500/500 [02:58<00:00, 2.81it/s]Plotting Metrics
Now that we have multiple performance metrics for each featurisation approach, rather than using a table, we can produce a scatter plot with a metric on each axis. Plots visualise differences between models and highlight instances where one metric might provide a misleading representation of performance.
Scatter plot
plt.figure(figsize=(10, 6))
labels = {'ecfps': 'ECFP$4_{1024}$','pdv': 'PDV', 'func_fps': 'FCFP$4_{1024}$'}
for method, scores in results.items():
x = scores['mae'].mean()
# mae confidence intervals
x_ci =stats.t.interval(
0.95,
total_splits-1,
loc=x,
scale=stats.sem(scores['mae'])
)
y = scores['r2'].mean()
# r2 confidence intervals
y_ci =stats.t.interval(
0.95,
total_splits-1,
loc=y,
scale=stats.sem(scores['r2'])
)
# plot means
plt.scatter(x, y, alpha=0.5, label=labels[method])
# plot confidence intervals
plt.errorbar(x, y, alpha=0.5, xerr=(x_ci[1]-x_ci[0])/2, yerr=(y_ci[1]-y_ci[0])/2, capsize=5)
plt.title('Coefficent of determination versus mean absolute error on hPPB task')
plt.xlabel('MAE\u2193 [log$_{10}$(%)]')
plt.ylabel('R$^{2}\u2191$')
plt.legend()
plt.grid(True)
plt.show()
The legibility of a results section is an aspect of research that is often overlooked. Simple plots, such as the one above, help to communicate differences between approaches. Aggregated tables with results for many models on multiple benchmarks are much harder to read by comparison.
Statistical Tests
So, we’ve visualised our results, but are the approaches significantly different in performance? We can use t-tests, Tukey’s HSD tests, and analysis of variation (ANOVA) to evaluate performance differences between methods.
t-tests
A t-test is a method for testing whether there is a significant difference between groups of data. Here, we use paired t-tests, as test splits were the same for each model. Our null hypothesis is that there is no difference in metrics between LGBMs with PDVs and ECFPs. A t-test can be run in Python using the stats module in the scipy library:
MAE t-test
# MAE t-test between physicochemical descriptor vectors and extended-connectivity fingerprints
mae_ttest = stats.ttest_rel(results['pdv']['mae'], results['ecfps']['mae'])
f't-statistic = {mae_ttest[0]:.3}; p-value = {mae_ttest[1]:.3}; degrees of freedom = {mae_ttest.df}''t-statistic = -28.5; p-value = 1.16e-106; degrees of freedom = 499'How do we interpret this result? As the t-statistic is 
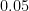

R2 t-test
# R2 t-test between physicochemical descriptor vectors and extended-connectivity fingerprints
r2_ttest = stats.ttest_rel(results['pdv']['r2'], results['ecfps']['r2'])
f't-statistic = {r2_ttest[0]:.3}; p-value = {r2_ttest[1]:.3}; degrees of freedom = {r2_ttest.df}''t-statistic = 22.9; p-value = 5.83e-80; degrees of freedom = 499'For 
Tukey HSD
So, a paired t-test is perfect for comparing two different models. For handling three or more models, we can compare methods in a pairwise manner using Tukey’s HSD test, which is another method for evaluating difference between means. This can be done with scipy:
MAE Tukey HSD
# MAE Tukey HSD
cols = ['PDV', 'ECFP4', 'FCFP4']
indexes = cols
cols = pd.MultiIndex.from_product([cols, ['statistic', 'p-value']])
mae_tukey = stats.tukey_hsd(results['pdv']['mae'], results['ecfps']['mae'], results['func_fps']['mae'])
tukey_mae_stats = pd.DataFrame(columns=cols, index=indexes)
tukey_mae_stats.loc[:, pd.IndexSlice[:, 'statistic']] = mae_tukey.statistic
tukey_mae_stats.loc[:, pd.IndexSlice[:, 'p-value']] = mae_tukey.pvalue
tukey_mae_stats| PDV | ECFP4 | FCFP4 | ||||
|---|---|---|---|---|---|---|
| statistic | p-value | statistic | p-value | statistic | p-value | |
| PDV | 0.0 | 1.0 | -0.103552 | 0.0 | -0.07745 | 0.0 |
| ECFP4 | 0.103552 | 0.0 | 0.0 | 1.0 | 0.026102 | 0.0 |
| FCFP4 | 0.07745 | 0.0 | -0.026102 | 0.0 | 0.0 | 1.0 |
Note that for these functions (t-tests, Tukey HSD, etc.) there are instances where the output p-value is 0; this is likely due to precision limits for float values. When the output p-value = 0 occurs, I suggest reporting the p-value as p-value < 0.001.
To visualise this kind of pairwise comparison, Ash et al. (2024) suggest using a Multiple Comparisons Similarity (MCSim) plots (i.e., heatmaps):
MCSim plot
plt.figure(figsize=(6, 5))
sns.heatmap(
mae_tukey.statistic, # values to plot
annot=True,
cmap='coolwarm',
center=0,
fmt=".2f",
cbar_kws={'label': 'Effect size'},
xticklabels=indexes, yticklabels=indexes, # label rows and cols with models
vmin=-0.2, vmax=0.2, # colour bar range
)
plt.show()
Analysis of variation (ANOVA) is a method for testing the difference in means between groups of data. Like t-tests and Tukey HSD, one-way ANOVA can be performed using the scipy library in Python:
One-way ANOVA
cols = pd.MultiIndex.from_product([indexes, ['F-statistic', 'p-value']])
anova_df = pd.DataFrame(columns=cols, index=indexes)
name_map = {'pdv':'PDV', 'ecfps':'ECFP4', 'func_fps': 'FCFP4'}
for i in results:
for j in results:
anova = stats.f_oneway(results[i]['r2'], results[j]['r2'])
anova_df.loc[name_map[j], (name_map[i], 'F-statistic')] = anova.statistic
anova_df.loc[name_map[j], (name_map[i], 'p-value')] = anova.pvalue
anova_df| PDV | ECFP4 | FCFP4 | ||||
|---|---|---|---|---|---|---|
| F-statistic | p-value | F-statistic | p-value | F-statistic | p-value | |
| PDV | 0.0 | 1.0 | 278.151946 | 0.0 | 174.92284 | 0.0 |
| ECFP4 | 278.151946 | 0.0 | 0.0 | 1.0 | 6.762882 | 0.009445 |
| FCFP4 | 174.92284 | 0.0 | 6.762882 | 0.009445 | 0.0 | 1.0 |
Now we have another indicator of the difference between means, the F-statistic, and p-values to determine whether the differences are significant. Let’s heatmap the p-values of these results:
Heatmap of p-values
pvals = anova_df.loc[:, pd.IndexSlice[:, 'p-value']].astype(float).to_numpy()
arr = np.zeros(pvals.shape, dtype=int)
arr[np.where(pvals < 0.05)] +=1
arr[np.where(pvals < 0.01)] +=1
arr[np.where(pvals < 0.001)] +=1
plt.figure(figsize=(6, 5))
colors = ["#d3d3d3", "#90ee90", "#008000", "#004d00"] # Custom colors for 0, 1, 2, 3
cmap = ListedColormap(colors)
ax = sns.heatmap(
arr, # values to plot
cmap=cmap,
xticklabels=indexes, yticklabels=indexes, # label rows and cols with models
cbar = True, square=True, fmt = 'd',
vmin=-0.5, vmax=3.5
)
colorbar = ax.collections[0].colorbar
colorbar.set_ticks([0, 1, 2, 3,])
colorbar.set_ticklabels(['p-value > 0.05', 'p-value < 0.05', 'p-value < 0.01', 'p-value < 0.001'])
plt.show()
For each pair of methods, there is a significant difference between the mean values for the coefficient of determination at a p-value threshold of 0.05.
Final thoughts
Hopefully this exploration of covariate shift splitting, visualisation, and statistical testing is helpful! Ash et al. (2024) go into greater detail and suggest more potential approaches for validating and communicating differences between models, so do give that a read too. Ultimately, all of these suggestions are in service of clarity; if another method for displaying your results communicates a desired point well, include it. After all, a results section is only as good as its figures.