
In deep learning based compound generation models the metric of fraction of RDKit-valid compounds is ubiquitous, but is problematic from the cheminformatics viewpoint as a large fraction may be driven by pyrrolic nitrogens (see below) rather than Texas carbons (carbon with 5 bonds like the Star of Texas). In RDKit, no error is more irksome that the KekulizeException or ValenceException from RDKit sanitisation. These are raised when the molecule is not correct. This would make the RDKit-valid a good metric, except for a small detail: the validity is as interpreted from the the stated implicit and explicit hydrogens and formal charges on the atoms, which most models do not assign. Therefore, a compound may not be RDKit-valid because it is actually impossible, like a Texas carbon, but in many cases it is because the formal charge or implicit hydrogen numbers of some atoms are incorrect. In both case, the major culprit is nitrogen. Herein I go through what they are and how to fix them, with a focus on aromatic nitrogens.
Pyrrolic and pyridinic nitrogen
The protonation of aromatic nitrogens is one cause of issue. Two configurations of nitrogen in arenes can be seen: one as an amine with no double bonds, called a pyrrolic nitrogen, and one as imine, with a double bond, called a pyridinic nitrogen. The former requires a hydrogen if not substituted, and this makes many compounds RDKit invalid.

In RDKit, a pyrrolic nitrogen needs a pendant group or an explicit hydrogen to be valid (the SMILES pattern ‘c1cncc1‘ is invalid, while ‘c1c[nH]cc1‘ is valid). As a result, the pictures of generated compounds in most diffusion papers do not feature unsubstituted pyrrolic nitrogens, but do feature substituted pyrrolic nitrogens, including bridgehead nitrogens, and pyridinic nitrogens do.
Having established that pyrrolic nitrogen can cause RDKit-invalidity, there is no simple pattern that can be used to spot pyrrolic nitrogens. For example, the SMARTS pattern '[nH0]' matches aromatic nitrogen with no hydrogens, which includes valid pyridinic nitrogens, while '[nH0r5]' will match aromatic nitrogen with no hydrogens on five-membered rings, which is a misunderstanding because you can get a pyridinic nitrogen on a five membered ring if there is a second nitrogen or an oxygen, and you can get a pyrrolic nitrogen on a six membered ring if there is another lone pair or pendant double bond. For Fragmenstein, I made a module, molecular-rectifier, to fix issues, including the pyrrolic nitrogen problem, but inelegantly by brute force —see below for a example. Before given a function to fix it, I need to go over charge and mention antiaromaticity.
To explain why there is not an easy fix let’s take a step back, all the way to an early chapter of Clayden, everyone’s favourite first year textbook. An aromatic compound, by Hückel’s rule, has (4n+2) π-bonded electrons, including lone pairs. Benzene has 6, but so have pyrrole, pyridine, but also fulvene or pyridone as discussed below. Not everything that you can draw with alternating double bonds is aromatic, as you can get antiaromatic compounds (4n π-bonded electrons) that are buckled, like cyclooctatetraene. Antiaromatic compounds are less stable and non-planar. Confusingly, a lowercase element in a SMARTS pattern for an antiaromatic compound will be accepted, but it will not be considered aromatic. Antiaromatic compounds are not planar, but AllChem.SanitizeMol will not check that, so a planar antiaromatic compound is rdkit-valid, but incorrect.
from rdkit import Chem
from rdkit.Chem import AllChem
mol = Chem.MolFromSmiles('c1ccccccc1') # COT but as aromatic
AllChem.SanitizeMol(mol)
print( all([bond.GetIsConjugated() for bond in mol.GetBonds()]) )
# True: it's conjugated
print( len(mol.GetAromaticAtoms()) )
# 0 not 8: it's not aromatic
print( Chem.MolToSmiles(mol) )
# 'C1=CC=CC=CC=C1'
Oxygen has two lone pairs, but only one is delocalised, so furan is aromatic like a pyrrole. A positive charge, due to a donor bond between a lone pair and a proton does not matter, this means that pyridinium is still aromatic. In the case of pendant carbonyls / alkenes, the trick is drawing in Kekulé form the right resonance structure. In the case of lactams, as seen in pyridone, drawing it in lactim form makes it look like pyridine. This also means Caffeine is aromatic… although caffenic acid is the aroma compound of coffee —yup, bad jokes are my specialty.
A nice insight into why kekulisation is tricky comes from the ‘three’ isomers of pyridone. Pyridone is an aromatic 6-membered ring with a lactam. However, whereas 2-pyridone and 4-pyridone can be draw in Kekulé form, ‘3-pyridone’ cannot adopt the lactam, so adopts the lactim form (3-hydroxypyridine) or the zwitterion form (pyridin-1-ium-3-olate). Therefore, 3-pyridone requires bonds changed so we can truly stamp it with the RDKit-invalid stamp.
Valence error due to charge
The valency of an atom is driven by how many shareable electrons the element has, and when the atom is uncharged it is super-simple (3 single bonds for boron, 4 for carbon, 3 for nitrogen and 2 for oxygen, sulfur can have 2 single bonds or 2 double and 2 single when hypervalent, hypervalent phosphorous 3 single and 1 double). When a lone pair on an atom forms a dative bond with another atom, you have a positive formal charge. Carbon has no lone pairs, so Texas carbon is impossible —in a SN2 transition state, you have partial bonds. While, for oxygen, oxocarbenium is only in transition states. Nitrogen however is very happy donate its lone pair, including when delocalised in aromatic compounds, such as methylpyridinium.
Here is a snippet to fix missing charges on nitrogens in a non-aromatic compound. When a SDF file lacks charge as is common in the catalogues of certain vendors, so that example is taken.
from rdkit import Chem, RDLogger
from rdkit.Chem import AllChem
from rdkit.Chem.SaltRemover import SaltRemover
path: Path = ...
saltremover = SaltRemover() # Enamine used to have salts explicitly in some compound SMILES
def add_nitrogen_charges(mol: Chem.Mol):
mol.UpdatePropertyCache(strict=False)
ps = Chem.DetectChemistryProblems(mol)
if not ps:
Chem.SanitizeMol(mol)
return mol
for p in ps:
if p.GetType()=='AtomValenceException':
at: Chem.Atom = mol.GetAtomWithIdx(p.GetAtomIdx())
if at.GetAtomicNum()==7 and at.GetFormalCharge()==0 and at.GetExplicitValence()==4:
at.SetFormalCharge(1)
Chem.SanitizeMol(mol)
return mol
# sdf catalogues such as MCule or Life Chemicals are sdf.gz
with gzip.open(path) as gzh:
with Chem.ForwardSDMolSupplier(gzh, sanitize=False) as sdfh:
RDLogger.DisableLog('rdApp.*')
for mol in sdfh:
if mol is None:
continue
mol = saltremover.StripMol(add_nitrogen_charges(mol))
Chem.SanitizeMol(mol) # yay!
RDLogger.EnableLog('rdApp.*')
Parenthetically, albeit beyond the scope of this post, pH specific charge can be added. during follow-up compound design, a common mistake folk make is forgetting that the nitrogens on piperidines, piperazines and morpholinos are actual charged —openbabel can automatic fix by pH (obmol.CorrectForPH(pH)), but not RDKit. Here is a snippet to go from RDKit and back. The main method is obmol.CorrectForPH(pH). OpenBabel has another method that is helpful, namely obmol.PerceiveBondOrders(), which adds bond order based on angles.
from openbabel import openbabel as ob
def convert_perceived_bonds_pH(mol: Chem.Mol, pH: float=7.4) -> Chem.Mol:
"""
Add bond order and pH to a rdkit molecule
:param mol:
:param pH:
:return: different Chem.Mol returned
"""
obmol = ob.OBMol()
conv = ob.OBConversion()
conv.SetInFormat("pdb")
if not mol.HasProp('_Name'):
mol.SetProp('_Name', 'molecule')
conv.ReadString(obmol, Chem.MolToPDBBlock(mol))
# obmol.PerceiveBondOrders()
obmol.CorrectForPH(pH)
conv.SetOutFormat("mol")
neomol: Chem.Mol = Chem.MolFromMolBlock(conv.WriteString(obmol).strip())
for k, v in mol.GetPropsAsDict().items():
if isinstance(v, bool):
neomol.SetBoolProp(k, v)
elif isinstance(v, int):
neomol.SetIntProp(k, v)
elif isinstance(v, float):
neomol.SetDoubleProp(k, v)
else:
neomol.SetProp(k, v)
return neomol
Bond order
The deep learning algorithms assign bond order, so I am operating that these should not be altered, whereas formal charges and protonation is fair game. However, it is worth discussing.
In RDKit, one can also perceive bond order from unconnected atoms with the submodule rdDetermineBonds. This is not to be confused with AllChem.AssignBondOrdersFromTemplate which requires a reference compound.
from rdkit import Chem from rdkit.Chem import AllChem from rdkit.Chem import rdDetermineBonds mol = ... # atom cloud assert mol.GetNumConformers(), 'Molecules lacks coordinates' rdDetermineBonds.DetermineBonds(mol) # perceive connections and bond order # or `rdDetermineBonds.DetermineConnectivity(mol)` and then `rdDetermineBonds.DetermineBondOrders(mol)` # for connectivity, the roundtrip `Chem.MolFromPDBFile(Chem.MolToPDBBlock(mol), proximityBonding=True)` will do it.
The Hoogeboom et al. 2022 EDM does it by bond length which is unusual as far as I know, but there may be a reason for this.
I have not compared the three approaches head-to-head on CCDC compounds, so I cannot say which is best. However, these have different relationships with charge.
Enforced conjugation by added charges
In some cases, in enforcing a ring to be conjugated (i.e. having alternating single / double bonds with occasional lone pairs) to make it aromatic, one could conceive an aromatic carbanion, but these are generally unstable.
Now to make things complicated. Mesoionic compounds, like sydnone, are curious as they lack an uncharged resonance form. They are aromatic with a charged-pyridinic nitrogen and a carbanion (or hydroxylate), making them especially tricky to get the SMILES right. This group features in mesocarb (Sydnocarb), a soviet amphetamine-like compound, and in some lead compounds, so is not fully a curiosity like compounds that look fun like penguinone. they are uncommon in catalogues, for example Enamine and Mcule do not have them (I think because the N-substituent has to be present before the ring is closed, preventing a series of adaptable simple building blocks). Nevertheless, sydnone is real and is RDKit-valid if written correctly.
Bodipy
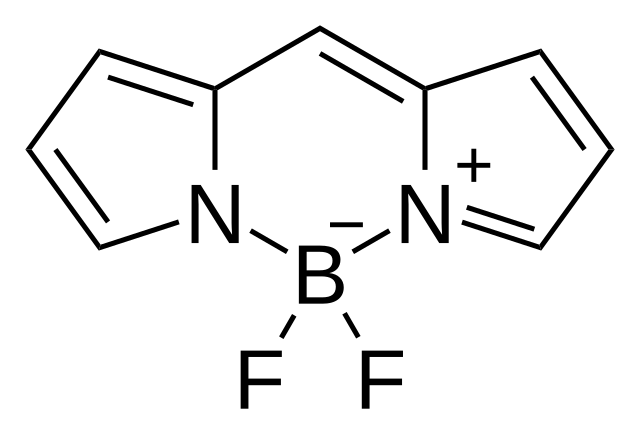
A special case in valence errors due to charge may arise with tetravalent boron. This boron anion features in bodipy dyes, so depending on the training dataset, these could appear a lot. azabora-arenes are possible, but are mainly uncommon catalysts, with some exceptions like diazaborine B. Boron is not in the MMFF94 forcefield, so further issues may happen. Also, as mentioned in a previous post, boronic acid/ester is a reactive group for certain reactions, so a medchemist / reviewer will not be happy with a boronic acid unless its a reversible warhead. This all moot as it depends if boron is an acceptable element in training the model, which I do not believe is the case in any model.
Radicals
I love the madness of biochemical anti-markovnikov reactions in radical SAM enzymes, but radicals are best avoided. Radicals are unstable in most cases, even if they are RDKit valid (see caveat about stability below).
Radicals are a problem in certain cases such as when the explicit number of hydrogens is zero and implicit hydrogens are forbidden. This, for non-arenes, can be reversed via Chem.Atom.SetNoImplicit.
atom.SetNoImplicit(False)
mol = Chem.MolFromSmiles('[CH2-]') # CH3- is correct...
atom: Chem.Atom
for atom in mol.GetAtoms():
atom.SetNumRadicalElectrons(0)
atom.SetNumExplicitHs(0)
atom.SetNoImplicit(False)
AllChem.SanitizeMol(mol)
mol
Alternatively, to be explicit about it:
mol = Chem.MolFromSmiles('[CH2-]')
periodic: Chem.PeriodicTable = Chem.GetPeriodicTable()
atom: Chem.Atom
for atom in mol.GetAtoms():
if atom.GetNumRadicalElectrons():
atom.SetNumRadicalElectrons(0)
expected_Hs: int = atom.GetNumExplicitHs() + (periodic.GetDefaultValence(atom.GetSymbol()) - atom.GetExplicitValence() + atom.GetFormalCharge())
atom.SetNumExplicitHs(expected_Hs)
atom.UpdatePropertyCache()
AllChem.SanitizeMol(mol)
mol
This will not work on arenes such as ‘c1cncc1‘.
Fixing the kekulisation and valence errors only
As mentioned, I used my molecular-rectifier module, an over-zelous rule-based algorithm which fixes RDKit-invalid molecules beyond the issues stated. For example a texas carbon will become a RDKit-valid sulfone as the next step is a catalogue search. It also fixes odd molecules such as benzo-cyclopropane (which are RDKit-valid, but a product of poor connectivity choices) and absorbs atoms that are too close. So this is not usable for a fair use of the RDKit-valid metric on multiple fronts.
from molecular_rectifier import Rectifier mol = ... recto = Rectifier(mol, valence_correction='charge') recto.fix_issues() # fixes valence issues etc. #recto.fix() # this corrects some problems beyond RDKit-validity recto.mol
Here are some compounds, that should and should not be redeemed by correcting charge and implicit hydrogens, but leaving element identity and bond order intact.
# Irredeemable
texas = Chem.MolFromSmiles('C(C)(C)(C)(C)(C)', sanitize=False) # irredeemable but will be fixed as a weird sulfone?!
# Redeemable
quat_amine = Chem.MolFromSmiles('N(C)(C)(C)(C)', sanitize=False) # redeemable --> '[N+](C)(C)(C)(C)'
pyrrole = Chem.MolFromSmiles('c1ccnc1', sanitize=False) # redeemable --> 'c1cc[nH]c1'
imidazole = Chem.MolFromSmiles('c1ncnc1', sanitize=False) # redeemable --> 'c1cc[nH]c1'
pyridinium = Chem.MolFromSmiles('c1cccn(C)c1', sanitize=False) # redeemable --> 'c1ccc[n+](C)c1'
pyridone = Chem.MolFromSmiles('c1ccc(=O)nc1', sanitize=False) # redeemable --> 'c1ccc(=O)[nH]c1'
isooxadiazole = Chem.MolFromSmiles('n1(cccc2)c2cno1', sanitize=False) # redeemable --> 'n1(cccc2)c2c[nH]o1'
sydnone = Chem.MolFromSmiles('n1oc(=O)cn1C', sanitize=False) # redeemable --> 'n1oc(=O)[cH1-][n+]1C'
The general principle behind the rectifier still counts. Although, to do things properly things get complicated for the fused ring, isooxadiazole.
# Main function: ``fix_kekulisation``.
import itertools
from typing import List, Sequence, Union
from rdkit import Chem, RDLogger
def strip_radicals(mol: Chem.Mol):
"""clear prior explicit Hs and radicals"""
for atom in mol.GetAtoms():
atom.SetNoImplicit(False)
atom.SetNumRadicalElectrons(0)
atom.SetNumExplicitHs(0)
atom.UpdatePropertyCache()
def extend_conjugation(mol, bad_idxs: Sequence[int]) -> List[int]:
"""
Extend the indices to include all conjugations
I honestly don't know if there's a function in RDKit that does this.
Test:
.. code-block:: python
cy3 = Chem.MolFromSmiles('C(=CC([H])=C1Cc2ccccc2N1C)C1=[N+](C)c2ccccc2C1')
extend_conjugation(cy3, [6])
"""
changed = False
bad_idxs = list(bad_idxs)
bad_atoms = [mol.GetAtomWithIdx(i) for i in bad_idxs]
atom: Chem.Atom
neigh: Chem.Atom
new_bad = []
for atom in bad_atoms:
for neigh in atom.GetNeighbors():
ni: int = neigh.GetIdx()
if ni in bad_idxs:
continue
elif neigh.GetIsAromatic():
bad_idxs.append(ni)
new_bad.append(neigh)
changed = True
elif mol.GetBondBetweenAtoms(atom.GetIdx(), ni).GetIsConjugated():
# pendant
new_bad.append(neigh)
bad_idxs.append(ni)
changed = True
bad_atoms.extend(new_bad)
return bad_idxs if not changed else extend_conjugation(mol, bad_idxs)
def test_protonation(mol: Chem.Mol,
idxs: Sequence[int],
problematics: Sequence[int]) -> Union[Chem.Mol, None]:
copy = Chem.Mol(mol)
for idx in idxs:
atom: Chem.Atom = copy.GetAtomWithIdx(idx)
if atom.GetDegree() == 2: # pyrrolic N?
atom.SetNumExplicitHs(1)
elif atom.GetDegree() == 3: # pyridinium-like N?
atom.SetFormalCharge(+1)
else:
pass
problems = Chem.DetectChemistryProblems(copy)
if not any([problem.GetType() == 'KekulizeException' for problem in problems]):
return copy
elif not any([problematics in p.GetAtomIndices() for p in Chem.DetectChemistryProblems(copy)]):
return copy
else:
return None
def create_combos(all_atoms) -> List[List[int]]:
"""
Returns a list of list with single possible pyrrolic N first, the pyridinium, then combos.
"""
combos: List[List[Chem.Atom]] = [atoms for r in range(1,len(all_atoms)+1) for atoms in itertools.combinations(all_atoms, r=r)]
combos = sorted(combos, key=lambda atoms: sum([atom.GetDegree() for atom in atoms]))
return [[atom.GetIdx() for atom in atoms] for atoms in combos]
def fix_kekulisation(mol: Chem.Mol, leave_idxs=()):
"""
This corrects protonation/charge problems on arenes due to nitrogen.
It simply gets the atoms that fail kekulisation, expands to the full conjugated system,
goes through the nitrogens trying the ones with two neighbours as pyrrolic N,
and the ones with three as pyridinium-like.
This is not perfect: protonation/charge issues on more complicated cases with carbanions etc. will be assigned incorrectly.
Also, does not care if aromatic or antiaromatic.
The combinations are done first single pyrrolic N, then single pyridinium-like,
double pyrrolic N etc. See ``create_combos``.
"""
RDLogger.DisableLog('rdApp.*')
problems = Chem.DetectChemistryProblems(mol)
if len(problems) == 0: # success!
AllChem.SanitizeMol(mol)
RDLogger.EnableLog('rdApp.*')
return mol
for problem in problems:
if problem.GetType() == 'KekulizeException':
problematics: List[int] = p.GetAtomIndices()
bad_atoms: List[Chem.Atom] = [mol.GetAtomWithIdx(i) for i in extend_conjugation(mol, problematics)]
bad_nitrogens: List[Chem.Atom] = [atom for atom in bad_atoms if atom.GetSymbol() == 'N']
combos = create_combos(bad_nitrogens)
for atoms in combos:
if leave_idxs == atoms:
continue # prevents recursion...
tested = test_protonation(mol, atoms, problematics)
if tested:
return fix_kekulisation(tested, atoms)
raise ValueError('failed to fix', problems)
Caveat one: Not just to make reviewers happy
Sure, reviewers like the RDKit-valid metric, so do keep using it… but not only for appeasement: just like a t-test it has limits and merits. Herein, I just want to point out its limits and its merits in order to endow a better picture.
Limits:
- Charge and protonation have nothing to do with most deep learning algorithms, so in my view these should not be failures, but fixed beforehand.
- It does not assess strain: an antiaromatic compound could be masqueraded as aromatically planar
- RDKit validity is not stability (see caveat 2)
Merits:
- It rejects compounds that have a forbidden degree of bonding
- It is easy to use
Caveat two: RDKit-invalid ≠ unstable ≠ impossible ≠ unmakeable
A compound that passes RDKit.Chem.rdmolops.SanitizeMol is not necessarily stable. Stability depends on solvent, pH and temperature. A transition state in a reaction can be encoded as a rdkit.Chem.Mol, but it is not stable. In addition to the conditions driving stability, there are technical nuances like protonation and tautomerisations: acetic acid in pH 7 water is ‘unstable’ as it deprotonates to acetate. And even when a compound is not stable, there is a range of stability. In this vein (i.e. energy profiles), whereas a truly impossible compound violates the laws of chemistry, there are many compounds that are so strained that they are not synthesisable under most conditions. For example, Bredt’s rule (rule = trend) states that you cannot have a bridgehead atom with double bond, yet, in typical synthetic chemistry fashion, folk have hunted hard and managed to make anti-Bredt compounds. These are more of a case of ‘hard-to-synthesise compounds‘. Catalysis is all about changing the energy profile by lowering the transition state peak and making the products less energetic that the reactants. Strain release chemistry uses strained compounds like propellanes to achieve unusual chemistry, which is less strained that the reactants, that is to say, strain is not a present/absent property, but a range of energies. Predicting if a scaffold or group is unstable is non trivial, but a very crude method is to check in NIH:Cactus with the ‘Names’ setting to see if it exists and if it has a common name: Say I want the carbon version of sulfone, so a hypervalent sulfur with two double bonded carbons and two single bonded carbon substituents (see figure), when I look this up nothing comes up as it will tautomerise / hydrolyse and is hard to make —parenthetically this is a shame as the theoretical weird world of thia-based Mobius-type spiro-aromatics could be mighty useful (for a ref in the deep end see this).

Hypervalent sulfur is generally fine —saccharin features an aromatic sulfur (VI) with dioxide pendants. A simple universal case of correct by RDKit but not stability would be hypervalent halogens. Chlorine has generally a valency of 1, but can be hypervalent: chlorine(IV) dioxide (used as an industrial bleach) has valence of 4. A hypothetical hypervalent-chlora-arene would be unutterably unstable, but as nothing in the training datasets will have a hypervalent chloride these should not happen (SMARTS pattern: ‘[Cl,Br,I;X2,X3,X4]‘).
In short, the set of RDKit-invalid compounds, unstable compounds, impossible compounds and unmakable compounds partially overlap, but none are not full subsets of the other.
Atom types in molecular mechanics
Parenthetically, it is worth mentioning that pyridinic vs. pyrrolic nitrogens is a problem in molecular mechanics too, which gets resolved via specific atom types. Specifically, therein atom types are not simply element symbols, but have also aromaticity, hybridisation state and other differentiators (like pyridinic, pyrrolic or pyridiniyum-like), leading to well over 50 atom types in most forcefields…
Conclusion
The RDKit-valid metric is a useful metric to eliminate heinous compounds, such as Texas carbon/nitrogen/oxygen or their relatives carba-spiro-arenes, which can easily occur in an algorithm struggling with tetrahedral geometry. However many compounds can be made valid by correcting, charge and implicit hydrogens, as discussed above.