
The ability to successfully apply previously acquired knowledge to novel and unfamiliar situations is one of the main hallmarks of successful learning and general intelligence. This capability to effectively generalise is amongst the most desirable properties a prediction model (or a mind, for that matter) can have.
In supervised machine learning, the standard way to evaluate the generalisation power of a prediction model for a given task is to randomly split the whole available data set 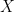
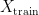
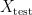
Since in this scenario the model has never seen any of the examples in 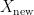
No.
In practise, one can regularly observe a situation where a machine learning model which performs well on a randomly selected test set
If the data split for the initial data set
Unfortunately, a random uniform data split is rarely a good simulation of practical reality where a newly collected data set
To get a more reliable picture of the real-world predictive capabilities of a trained machine learning model one must find a way to model a meaningful distributional shift and build it into the test set
Measuring out-of-distribution generalisation is of particular relevance in the field of molecular property prediction where distributional shifts tend to be large and difficult to handle for machine learning models. Different molecular data sets obtained by distinct pharmaceutical companies and research groups often contain compounds from vastly different areas of chemical space that exhibit high structural heterogeneity. An elegant solution for the modelling of such distributional shifts in chemical space is given by the idea of scaffold splitting.
The notion of a (two-dimensional) molecular scaffold is described in the article by Bemis and Murcko [2]. A molecular scaffold reduces the chemical structure of a compound to its core components, essentially by removing all side chains and only keeping ring systems and parts which link together ring systems. An additional option for making molecular scaffolds even more general is to “forget” the identities of the bonds and atoms by replacing all atoms with carbons and all bonds with single bonds.
Bemis-Murcko scaffolds can be automatically generated in RDKit via the following Python code:
# how to extract the Bemis-Murcko scaffold of a molecular compound via RDKit # import packages from rdkit import Chem from rdkit.Chem.Scaffolds import MurckoScaffold # define compound via its SMILES string smiles = "CN1CCCCC1CCN2C3=CC=CC=C3SC4=C2C=C(C=C4)SC" # convert SMILES string to RDKit mol object mol = Chem.MolFromSmiles(smiles) # create RDKit mol object corresponding to Bemis-Murcko scaffold of original compound mol_scaffold = MurckoScaffold.GetScaffoldForMol(mol) # make the scaffold generic by replacing all atoms with carbons and all bonds with single bonds mol_scaffold_generic = MurckoScaffold.MakeScaffoldGeneric(mol_scaffold) # convert the generic scaffold mol object back to a SMILES string format smiles_scaffold_generic = Chem.CanonSmiles(Chem.MolToSmiles(mol_scaffold_generic)) # display compound and its generic Bemis-Murcko scaffold display(mol) print(smiles) display(mol_scaffold_generic) print(smiles_scaffold_generic)

If we now have a molecular data set 
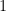
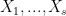
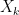
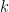

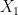

One appropriate way to now produce a scaffold split of the molecular data set 
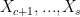

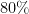
While a scaffold split is certainly not perfect, it is already a lot better than a uniform random split at providing a relevant measure of the practical utility of a molecular property prediction model. It mimics a situation where the training set
References:
[1] Poggio, Tomaso, and Christian R. Shelton. “On the mathematical foundations of learning.” American Mathematical Society 39.1 (2002): 1-49.
[2] Bemis, Guy W., and Mark A. Murcko. “The properties of known drugs. 1. Molecular frameworks.” Journal of medicinal chemistry 39.15 (1996): 2887-2893.