
When a ring flips, how do we calculate RMSD?
This surprisingly simple question leads to a very interesting problem! If we take a benzene molecule, say, and rotate it 180 degrees, then we have the exact same molecule, but if we have a data structure in which our atoms are labelled, and we apply the same transformation to the atomic positions, the numbering does not reflect that symmetry. If we were then naively to calculate the RMSD it would be huge, despite the fact that the molecule is, chemically speaking, identical.
How can we make our RMSD calculations reflect these symmetries?
We will look to combine graph theory, which can be used to model the structures of molecules, and group theory, which can be used to model their symmetries, in order to answer this question.
RMSDs
An RMSD between two structures is simply defined as:

Where 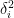
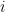
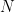
Graphs
A graph is an ordered pair, 
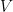
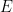
Groups
The mathematical theory that deals with symmetry is group theory, so it seems natural that this is where we would look for a solution.
Groups
To describe the symmetries of an object we need two things: a set of states and a set of transitions.
A group is a set, 

Associativity: For all 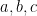
Identity: There exists an element, 
Inverse: For every element, 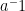
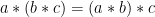
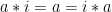
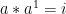
Group Action
Consider a group

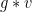
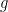

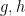
Identity:
Compatability:
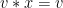
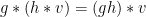
Orbits
Now that we have the elements of the group, we can see where each of them can move under the group action.
The Orbit of an element, 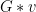
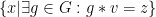
Isomorphisms and Automorphisms
An isomorphism is a structure-preserving (for some sense of the word) bijection from some mathematical object to another. When that other object is itself then isomorphism is called a automorphism.
Importantly, the set of all automorphisms satisfies the group axioms when the operation is function composition.
Automorphisms on graphs
For a graph, the structure we might be most interested in preserving is adjacency, so let us consisder all relabellings of the graph verticies that preserved adjacency: i.e. if an edge connects vertex 1 and vertex 2, then in the relabeling it must also.
These mappings do exist (although may only be the identity mapping), and together they form a group under composition (i.e. relabel according to one mapping, then by another afterwards). The action of this group on the graph is then the application of the relabellings.
Putting it together to find which atoms could match which atoms
The ideas above can be trivially extended to graphs which have some additional typing structure: in the case we are interested in, small molecules, it is that atom type. We can then look for those automorphisms that preserve the atom typed adjacency of the atoms in the molecule.
Python example code
Here is some example code using this technique. Some of this is borrowed from the excellent tutorial in the gemmi docs which I highly reccomend to anyone interested in working with crystalographic data.
import sys
import networkx
from networkx.algorithms import isomorphism
import gemmi
# Function to get graph from gemmi comp_chem object
def graph_from_chemcomp(cc):
G = networkx.Graph()
for atom in cc.atoms:
G.add_node(atom.id, Z=atom.el.atomic_number)
for bond in cc.rt.bonds:
G.add_edge(bond.id1.atom, bond.id2.atom)
return G
# Function to get rmsd from atom lists
def rmsd_from_atom_lists(atom_list_1, atom_list_2):
distances = []
for atom1, atom2 in zip(atom_list_1, atom_list_2):
pos1 = atom1.pos
pos2 = atom2.pos
distance = pos1.dist(pos2)
distances.append(distance)
return np.mean(distances)
# Function to calculate a symmeterised rmsd
def symmetrised_rmsd(mol1, mol2, symmetries):
rmsds = []
mol_1_atom_list = mol1.atoms
mol_2_atom_list = mol2.atoms
for symmetry in symmetries:
mol_2_atom_symmetry_list = [mol_2_atom_list[x] for x in symmetry.values()]
rmsd = rmsd_from_atom_lists()
rmsds.append(rmsd)
# Paths to smolecules
f1 = "/path/to/dock/1"
f2 = "/path/to/dock/2"
# Load in a small molecule
block = gemmi.cif.read(f1)[-1]
cc1 = gemmi.make_chemcomp_from_block(block)
# Load in a second
block = gemmi.cif.read(f2)[-1]
cc2 = gemmi.make_chemcomp_from_block(block)
# Restric to heavy atoms
cc1.remove_hydrogens()
cc2.remove_hydrogens()
# Get graphs
graph1 = graph_from_compchem(cc1)
graph2 = graph_from_compchem(cc2)
# Calculate the automorphisms
node_match = isomorphism.categorical_node_match('Z', 0)
symmetries = isomorphism.GraphMatcher(graph1, graph1, node_match=node_match)
# Get the rmsd
rmsd = symmetrised_rmsd(graph1, graph2, symmetries .iter_isomorphisms(isomorphisms_iter))
Going fast: symmetric motifs
There is a problem with the above code: it is slow. It checks every automorphism. But… that is much more work than we need to do!
This is because, small molecules often have independent symmetries i.e. imagine the three hydrogens in a methyl group can be permuted without needing to relabel the rest of the molecule.
We call such small regions “symmetric motifs”, and by finding the set of symmetric motifs, we could actually make claims such as: “this is the minimum number of atom distances you need to check in order to calculate a symmetrised RMSD”.
Of course, depending on the molecule and the number of RMSDs you want to calculate, that up-front effort of analysing the graph might not actually be worth it. Or it might save you 10’s of millions of calculations.