In last week’s journal club we delved into the history of Ramachandran plots (Half a century of Ramachandran plots; Carugo & Djinovic-Carugo, 2013).

Polypeptide backbone dihedral angles. Source: Wikimedia Commons, Bensaccount
50 years ago Gopalasamudram Narayana Ramachandran et al. predicted the theoretically possible conformations of a polypeptide backbone. The backbone confirmations can be described using three dihedral angles: ω, φ and ψ (shown to the right).
The first angle, ω, is restrained to either about 0° (cis) or about 180° (trans) due to the partial double bond character of the C-N bond. The φ and ψ angles are more interesting, and the Ramachandran plot of a protein is obtained by plotting φ/ψ angles of all residues in a scatter plot.
The original Ramachandran plot showed the allowed conformations of the model compound N-acetyl-L-alanine-methylamide using a hard-sphere atomic model to keep calculations simple. By using two different van der Waals radii for each element positions on the Ramachandran plot could be classified into either allowed regions, regions with moderate clashes and disallowed regions (see Figure 3 (a) in the paper).
The model compound does not take side chains into account, but it does assume that there is a side chain. The resulting Ramachandran plot therefore does not describe the possible φ/ψ angles for Glycine residues, where many more conformations are plausible. On the other end of the spectrum are Proline residues. These have a much more restricted range of possible φ/ψ angles. The φ/ψ distributions of GLY and PRO residues are therefore best described in their own Ramachandran plots (Figure 4 in the paper).
Over time the Ramachandran plot was improved in a number of ways. Instead of relying on theoretical calculations using a model compound, we can now rely on experimental observations by using high quality, hand picked data from the PDB. The way how the Ramachandran plot is calculated has also changed: It can now be seen as a two-dimensional, continuous probability distribution, and can be estimated using a full range of smoothing functions, kernel functions, Fourier series and other models.
The modern Ramachandran plot is much more resolved than the original plot. We now distinguish between a number of well-defined, different regions which correlate with secondary protein structure motifs.
Ramachandran plots are routinely used for structure validation. The inherent circular argument (A good structure does not violate the Ramachandran plot; The plot is obtained by looking at the dihedral angles of good structures) sounds more daring than it actually is. The plot has changed over time, so it is not as self-reinforcing as one might fear. The Ramachandran plot is also not the ultimate guideline. If a new structure is found that claims to violate the Ramachandran plot (which is based on a huge body of cumulative evidence), then this claim needs to be backed up by very good evidence. A low number of violations of the plot can usually be justified. The Ramachandran plot is a local measure. It therefore does not take into account that domains of a protein can exert a force on a few residues and just ‘crunch’ it into an unusual conformation.
The paper closes with a discussion of possible future applications and extensions, such as the distribution of a protein average φ/ψ and an appreciation of modern web-based software and databases that make use of or provide insightful analyses of Ramachandran plots.

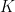
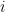
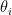
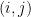
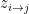
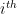
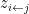

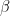

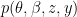
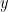
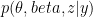

 ) that maximises the following posterior distribution for the number of bins:
) that maximises the following posterior distribution for the number of bins:
 is the number of bins,
is the number of bins,  is the data,
is the data, 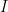 is prior knowledge about the problem, i.e. in particular the use of equal length bins and the range of data
is prior knowledge about the problem, i.e. in particular the use of equal length bins and the range of data 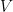 , which has the relation
, which has the relation  where
where  is the width of bins,
is the width of bins, 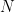 is the number of data points and
is the number of data points and 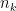 is the number of observations that fall in the
is the number of observations that fall in the 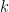 th
th  ) of the bins of the histogram is given by:
) of the bins of the histogram is given by: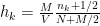 .
.



