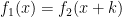Similarly to ‘words’, which need to be “assembled into sentences, paragraphs, chapters and books” to tell a story, ‘protein structures’ need to be assembled into protein complexes to perform a specific task. To form complexes, proteins interact with other proteins, DNA, RNA and small molecules using their interface residues. All those types of interactions are under intense scrutiny by the research community, each of them defining a distinct field of research. During my PhD I focused on protein-protein interactions (PPIs) and prediction of their interfaces. Modifications in PPIs affect the events that take place within cells which may lead to critical diseases such as cancer. Therefore, knowledge about PPIs and their resulting 3D complexes can provide key information for drug design.
Docking is a popular computational method which predicts the possible structure of the complex produced by two proteins using the known 3D structure of the individual proteins. However, docking of two proteins can result in a large number of different conformational models whose majority is far from correct. This highlights one of the main limitations of docking. Therefore, scoring functions have been proposed which are used to re-score and re-rank docked conformations in order to detect near-native models. One way to distinguish native-like models from false docked poses is to use knowledge of protein interfaces. If one knows the possible location of interface residues on each individual protein, docked complexes which do not involve those interfaces can be rejected. Therefore, accurate prediction of protein interfaces can assist with detection of native-like conformations.
Various methods have been proposed for predicting protein interfaces as mentioned above. A high number of methods investigate protein sequential or structural features in order to characterise protein interfaces. Usage of 3D structural properties has improved the sequence-based predictions. Moreover, evolutionary conservation was shown to be an important property. Therefore, methods have integrated various structural features along with evolutionary information to increase performance.
The combination of different features using various techniques has been investigated by intrinsic-based predictors. However, it seems that these methods have reached their saturation, and combination of more properties does not improve their prediction performance. On the other hand, many studies have investigated the 3D structure of binding sites among protein families. They discovered that the binding site localisation and structure are conserved among homologous. These properties have improved the detection of functional residues and protein-ligand binding sites. Therefore, predictors took advantage of structurally conserved residues among homologous proteins to improve binding site predictions.
Although homologous template-based predictors improve the predictions, they are limited to those proteins whose homologous structure exists. Therefore, methods have extended their search for templates to structural neighbours, since interface conservation exists even among remote structural neighbours. In addition, with the increase in experimentally determined 3D complexes good quality templates can be found for many proteins. Therefore, usage of structural neighbours is the current focus of template-based protein interfaces predictors.
Although, template-based methods are currently the predictors under the main focus, one of their main limitations is their dependency to availability of the QP 3D structure. Also, these predictors have not investigated the contribution of interacting partners of structural neighbours in the prediction. In addition, since these methods perform structural comparisons their computational time is high which limits their application to high-throughput predictions.
One of my PhD contributions was toward developing, T-PIP (Template based Protein Interface Prediction), a novel PIP approach based on homologous structural neighbours’ information. T-PIP addresses the above mentioned limitations by quantifying, first, homology between QP and its structural neighbours and, second, the diversity between the ligands of the structural neighbours (here, ligands refers to the interacting partners of proteins). Finally, predictions can be performed for sequences of unknown structure if that of a homologous protein is available. T-PIP’s main contribution is the weighted score assigned to each residue of QP, which takes into account not only the degree of similarity between structural neighbours, but also the nature of their interacting partners.
In addition, we used T-PIP prediction to re-rank docking conformations which resulted in T-PioDock (Template based Protein Interface prediction and protein interface Overlap for Docking model scoring), a complete framework for prediction of a complex 3D structure. T-PioDock supports the identification of near-native conformations from 3D models that docking software produced by scoring those models using binding interfaces predicted by T-PIP.

T-PioDock Pipeline
Exhaustive evaluation of interface predictors on standard benchmark datasets has confirmed the superiority of template base approaches and has showed that the T-PIP methodology performs best. Moreover, comparison between T-PioDock and other state-of-the-art scoring methods has revealed that the proposed approach outperforms all its competitors.
Accurate identification of near-native conformations remains a challenging task. Although availability of 3D complexes will benefit to template based methods such as T-PioDock, we have identified specific limitations which need to be addressed. First, docking software are still not able to produce native like models for every target. Second, current interface predictors do not explicitly refer to pair-wise residue interactions which leaves ambiguity when assessing quality of complex conformations.